遗传性出血性毛细血管扩张症一家系诊断与治疗研究
2017-02-23张树贤任玲徐静李祎
张树贤,任玲,徐静,李祎
(连云港市第一人民医院,江苏 连云港 222000)
遗传性出血性毛细血管扩张症一家系诊断与治疗研究
张树贤,任玲,徐静,李祎*
(连云港市第一人民医院,江苏 连云港 222000)
目的:探讨遗传性出血性毛细血管扩张症(HHT)家系的临床特征、诊断及治疗。方法:对先证者进行家系调查、体格检查及胃肠镜检查,经过沙利度胺治疗后复查胃肠镜。结果:本家系4代包括先证者在内共7 例患者。先证者经沙利度胺系统治疗后,症状缓解,胃镜复查出血灶明显减少。结论:遗传性出血性毛细血管扩张症家系以反复胃出血为主要表现,沙利度胺能够很好地控制患者的出血症状。
遗传性出血性毛细血管扩张症;家系调查;胃肠道出血;沙利度胺
遗传性出血性毛细血管扩张症(HHT),又名Osler-Rendu-Weber综合征,是一种常染色体显性遗传病。HHT以20~30岁为高发年龄,主要特征为皮肤、黏膜及内脏多发性毛细血管扩张并有出血倾向,临床表现取决于异常血管部位及范围,其发病率约1/5 000~1/8 000[1]。
1 家系资料
先证者陶某,女,70 岁,反复鼻出血40 年,病情渐加重伴乏力,因胃出血急诊入院。体格检查:重度贫血貌,营养中等,全身皮肤黏膜苍白,手指末端指腹可见点状出血点,浅表淋巴结未触及肿大,心肺未见异常。血常规:白细胞4.1×109/L,红细胞 2.02×1012/L,血红蛋白38 g/L,血小板222×109/L,凝血时间、凝血酶原时间均正常;其他检查未见异常改变。给予止血、输液、药物治疗及营养支持后患者病情好转,6 d后行胃镜和肠镜检查:胃体、胃窦可见多发边界清楚、点片状鲜红斑,十二指肠球部黏膜光滑,肠镜未见异常改变,提示胃毛细血管扩张症。病情稳定后出院,出院后给予沙利度胺口服治疗,每次25 mg,每日3次。后再次因胃出血入院。血常规:白细胞4.95×109/L,红细胞 3.05×1012/L,血红蛋白59 g/L,血小板207×109/L;其他检查未见异常改变。家系调查显示该患者所在家系共有4代包括先证者在内共7 例存在不同程度反复鼻出血、胃肠道出血。
患者胃镜检查示胃、十二指肠毛细血管扩张。追问病史患者家中共有兄妹5 人,患者为长女,其母有反复鼻出血和消化道出血史,已病故,其下2个妹妹有反复鼻出血史,不影响正常生活,均健在。否认近亲结婚。患者本人已婚,生育子女,其次女也有反复鼻出血史。结合患者病史、家族史和化验检查,根据Shovlin标准,此7 例家系成员均可确诊为HHT患者。
该HHT家系图谱见图1。胃镜图片见图2。
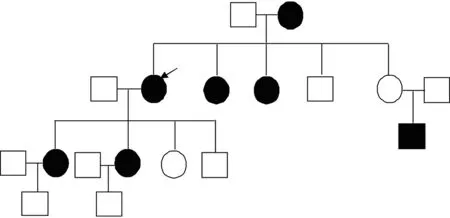
□正常男性,■患病男性,○正常女性,●患病女性,→先证者

图2 患者胃镜图片
2 诊断与治疗
2.1 临床诊断
目前HHT的临床诊断主要采用Shovlin标准[2]:第一,自发的、反复发作的鼻出血(夜间鼻出血更要高度考虑HHT);第二,多发性皮肤或黏膜的毛细血管扩张,如嘴唇、口腔、指甲、鼻;第三,内脏动静脉畸形( AVM),伴或不伴胃肠道出血的胃肠道毛细血管扩张、肺动静脉瘘、肝AVM、脑AVM、脊髓AVM;第四,家族史[1]。具备上述4 项中的3 项者可确诊;具备其中2 项者为疑诊;如果少于2 项,则HHT可能性不大。其中,胃肠道动静脉畸形约占44%,胃肠道出血的发病年龄偏高,一般在40~50 岁时出现。
2.2 基因突变与HHT
HHT是一种常染色体显性遗传疾病,目前已明确2 个染色体致病基因,即9号染色体的 ENG[3]和位于12号染色体的ALK-l突变基因[4]。由ENG基因突变所引起的HHT称为HHT1型;由ALK-l基因突变引起的HHT称为HHT2型,HHT2型较HTT1型更为常见。2004年Gallione等[5]发现了HTJP:在7个同时存在幼年息肉病( JP)和HHT (JP和HHT综合征)家族中,发现了位于染色体18q21的[SMAD4(MADH4)]基因突变,而未发现ENG及ALKI的突变。85%的HHT患者为ENG或ALK-1基因突变所致,目前,在HHT中ALK-1基因和ENG基因被鉴定出的突变类型大约有600余种,这些突变主要包括碱基缺失突变、碱基插入突变、错义突变及无义突变等[6]。近年研究发现转化生长因子-β(TGF-β)信号转导系统的异常及其他血管生长因子参与HHT发病过程[7]。HHT1型的肺动静脉畸形发生率较高,而HHT2型临床症状比较缓和,发病时间较晚,消化道累及率较高。
2.3 病理改变与HHT
HHT的病变部位在血管壁,表现为毛细血管扩张、内脏动静脉畸形和动脉瘤形成。主要的病理变化为部分小动脉、毛细血管、小静脉血管壁结构异常,血管壁变薄,有的部位仅由一层内皮细胞组成,外围包裹一层疏松结缔组织。由于其缺乏正常血管壁的弹力纤维及平滑肌成分,同时血管壁失去对交感神经和血管活性物质调节的反应能力,使得血管缺乏正常的舒缩功能。胃肠道出血常常发生在较大年龄人群中,胃肠道毛细血管扩张在胃肠道系统任何部位均可出现,发生率约为15%,最常受累部位是胃和十二指肠。
2.4 HHT的治疗
Sadick 等[8]研究表明HHT患者血清中血管内皮生长因子(VEGF)和TGF-β的水平升高。沙利度胺(反应停)最初在20世纪60年代应用于治疗妇女妊娠期间引起的呕吐反应,由于它对于胎儿的严重致畸作用(在胚胎形成的特定时期能够减少组织和器官的血液供应),使得反应停一度退出市场[9]。然而,近几年来沙利度胺又重新被用于治疗各种疾病,它能够很好地抑制胃肠道血管畸形所引起的出血[10],其具体的机制尚不明确,有报道称可能与调节血管生成的VEGF信号通路有关[11]。
由于HHT患者血清中VEGF的含量升高,国外也将VEGF的特异性抑制剂贝伐单抗应用于治疗HTT,抑制患者的出血症状[12]。贝伐单抗是一类重组的人单克隆抗体,能够特异性的抑制VEGF的活性,从而发挥抗出血作用。贝伐单抗在HHT患者中有一定的疗效,但是它也有一些不可避免的毒性作用[13],可能诱发心力衰竭、出血、动脉血栓事件、高血压危象以及肾病综合征等,加之其价格昂贵,患者很难接受,国内尚没有文献报道将贝伐单抗应用于HHT患者中。
3 讨 论
HHT为常染色体显性遗传性血管发育异常引起的疾病。1864年Sutton首次报道该病,1896年Rendu对该病进行了较详细的论述,1901年Osier报道该病的家族性及其临床特征,Weber也对该病进行了描述。因此,该病又被称为Osier-Rendu-Weber病[14]。
HHT的主要临床表现是身体某部位反复出血,最常见的出血部位为鼻腔黏膜,93%的患者均发生过鼻出血。HHT患者的皮肤、黏膜毛细血管扩张是局部反复出血的基础。典型的毛细血管扩张症病灶呈点状的、紧密交织的扩张的毛细血管丛,直径约1~4 mm。其他毛细血管扩张的常见部位包括消化系统、呼吸系统及泌尿生殖系统等[15]。本HHT家系患者的主要表现为反复胃肠道出血及继发性缺铁性贫血表现。
研究表明ENG基因和ALK-1基因在血管形成过程中起着重要作用,它们编码的蛋白质通过TGF-β超家族信号传导途径发挥作用[16]。在Smad依赖的信号途径中,配体和受体结合活化Ⅱ型TGF-β受体,使其发生磷酸化,引起Ⅰ型TGF-β受体磷酸化,使得受体相关的Smad 1、2、3、5、8活化,与Smad4结合,进入细胞核内进行转录、翻译,最终发挥效应。
HHT应与以下疾病相鉴别。第一,红痣:仅见于皮肤,鲜红色,高出皮肤,边缘清楚,指压不褪色。第二,蜘蛛痣:见于肝病及妊娠期妇女,用指尖或火柴头压迫蜘蛛痣中心,辐射状的小血管网即可褪色,很少出血。第三,小静脉扩张:常见于面部和大腿,多呈条状分布。第四,其他:消化道病变出血应注意与溃疡病、食道静脉曲张、消化道恶性肿瘤相鉴别,可通过病史、内窥镜检查观察黏膜毛细血管有无典型扩张等协助鉴别。
关于HHT的治疗,本病目前尚无特殊治疗方法,主要为对症治疗。鼻出血可用鼻腔填塞物或加压止血,严重反复的鼻出血或皮肤出血可采用激光凝固、冷冻、动脉栓塞或手术缝合等措施。胃肠道出血可用内镜下双频电切或激光技术处理。
沙利度胺因具有免疫调节及抗血管生成作用而被用于治疗多发性骨髓瘤、系统性红斑狼疮、克罗恩病及多种恶性肿瘤[17]。沙利度胺治疗HTT患者的皮肤黏膜出血及胃肠道血管扩张引起的出血疗效确切并且相对较安全,可以作为长期治疗的用药选择。
在临床中,当接诊以消化道出血为主要临床表现的患者时,应详细询问病史和体格检查,当遇到内镜下有胃肠道多发血管畸形时,应注意有无皮肤黏膜改变,要优先考虑是否有遗传性出血性疾病和全身性疾病,以尽量减少误诊和漏诊率。
[1]ZARRABEITIA R,ALBI ANA V,SALCEDO M,et al.A review on clinical management and pharmacological therapy on hereditary haemorrhagic telangiectasia (HHT)[J].Curt Vasc Pharmacol,2010,8 (4):473-481.
[2]SHOVLIN C L,GUTTMACHER A E,BUSCARINI E,et al.Diagnostic criteria for hereditary hemorrhagic telagi- ectasia(Rendu-Osler-Weber-syndrom)[J].Am J Med Genet,2000,91(1):66-67.
[3]HEUTINK P,HAITJEMA T,BREEDVELD G J,et al.Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity[J].J Med Genet,1994,31(12):933-936.
[4]JOHNSON D W,BERG J N,BALDWIN M A,et al.Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2[J].Nat Genet,1996,13(2):189-195.
[5]GALLIONE C J,REPETTO G M,LEGIUS E,et al.A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4)[J].Lancet,2004,363 (9 412):852-859.
[6]GOVANI F,SHOVLIN C.Hereditary haemorrhagic telangiectasia:a clinical and scientific review[J].Eur J Hum Genet,2009,17(7):860-871.
[7]GARCIA-TSAO G.Liver involvement in hereditary hemorrhagic telangiectasia(HHT)[J].J Hepatol,2007,46(3):499-507.
[8]SADICK H,RIEDEL F,NAIM R,et al.Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression[J].Haematologica,2005,90(6):818-828.
[9]THERAPONTOS C,ERSKINE L,GARDNER E R,et al.Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation[J].Proc Natl Acad Sci,2009,106(21):8 573-8 578.
[10]BAUDITZ J,SCHACHSCHAL G,WEDEL S,et al.Thalidomide for treatment of severe intestinal bleeding[J].Gut,2004,53(4):609-612.
[11]MELCHERT M,LIST A.The thalidomide saga[J].Int J Biochem Cell Biol,2007,39(7-8):1 489-1 499.
[12]DAVIDSON T M,OLITSKY S E,WEI J L.Hereditary hemorrhagic telangiectasia/avastin[J].Laryngoscope,2010,120(2):432-435.
[13]AMANZADA A,TOPPLER G J,CAMERON S,et al.A case report of a patient with hereditary hemorrhagic telangiectasia treated successively with thalidomide and bevacizumab[J].Case Rep Oncol,2010,3(3):463-470.
[14]GUTTMACHER A E,MARCHUK D A,WHITE R I J R.Hereditary hemorrhagic telangiectasia[J].N Engl J Med,1995,333(14):918-924.
[15]PAU H,CARNEY A S,MURTY G E.Hereditary hemorrhagic telangiesia (Osier-Weber-Rendu syndroume):otorhinolayngological manifestations[J].Clin Otolaryngol Allied Sci,2001,26(2):93-98.
[16]GUO X,WANG X F.Signaling cross-talk between TGF-beta/BMP and other pathways[J].Cell Res,2009,19(1):71-88.
[17]JACOBSON J M,GREENSPAN J S,SPRITZLER J,et al.Thalidomide for the treatment of oral aphthous ulcers in patients with human immunodeficiency virus infection.National institute of allergy and infectious diseases AIDS clinical trials group[J].N Engl J Med,1997,336(21):1 487-1 493.
(本文编辑: 张荣梅 )
The diagnosis and treatment of a familial disease-hereditary hemorrhagic telangiectasia
ZHANG Shuxian,REN Ling,XU Jing,LI Yi
(The First People′s Hospital of Lianyuangang,Lianyuangang 222000,China)
Objective:To learn more the clinical features,diagnosis and treatment of Hereditary hemorrhagic telangiectasia (HHT).Methods:The records before and after thalidomide therapy,including family investigation,physical examination and endoscopic examination,were reviewed of the patient who was presented with recurrent gastrointestinal bleeding,and later was diagnosed as HHT.Results:In the four-generation pedigree,including the proband himself,a total of 7 patients were suffered from HHT.After treatment by thalidomide,the patient′s gastrointestinal bleeding was effectively controlled.Conclusion:This family of HHT patients are characterized by repeated gastric bleeding,and thalidomide therapy is proved to be effective.
hereditary hemorrhagic telangiectasia; family survey; gastrointestinal bleeding; thalidomide
张树贤(1985— ),男,山东省聊城市人,硕士学位,医师,主要从事胃肠道疾病诊治工作。
1671-8631(2017)01-0013-04
R76
B
2016-05-12
*本文通讯作者:李祎
