纳米复合材料固定化酶的研究进展
2017-02-16相欣然黄和胡燚
相欣然黄和胡燚
综述
纳米复合材料固定化酶的研究进展
相欣然黄和胡燚*
(南京工业大学药学院,材料化学工程国家重点实验室,江苏先进生物与化学制造协同创新中心,南京210009)
载体材料的选择对固定化酶的性能有着至关重要的影响。纳米复合材料不仅具有纳米尺寸的特性,而且可以克服单一材料的不足,在固定化酶领域引起了广泛关注。本文就目前在固定化酶领域使用的纳米复合载体分类进行了系统的阐述,重点介绍了目前在固定化酶研究领域运用较为广泛的硅基纳米复合材料、碳基纳米复合材料和纳米纤维复合材料等材料的制备方法及不同材料对酶学性能的影响,并对这些纳米复合材料固定化酶发展前景进行了展望。
纳米复合材料;固定化酶;硅基材料;碳基材料
0 引言
酶是一种可以高效控制特定化学反应的一种通用的生物催化剂,在实际应用中通常将酶作固定化处理。固定化酶由于其高稳定性、可重用性、易分离等性质被广泛地应用于化学、生物、农业和医药等领域[1-3]。酶固定化方法主要包括物理吸附法、共价结合法、包埋法和交联法。制备固定化酶过程中所采用的固定化技术、载体、介质条件和所催化反应类别会在一定程度上导致酶失活、变性,从而使酶的催化性能或保留活性降低,其中载体所带来的分配效应、空间障碍效应和扩散限制效应是影响固定化酶催化效率的主要因素,因此探寻可行、有效的固定化载体来增强固定化酶的催化性能一直是固定化酶研究的热点[4-6]。
纳米尺度的材料由于具有特殊的表面效应、体积效应、量子尺寸效应和宏观量子隧道效应等,在声、光、电、磁、热性能方面呈现出了新的特性,在全球范围内引起了科学和产业界的极大关注[7]。与传统大尺寸材料相比,纳米材料还具有大比表面积、表面易于修饰、与酶分子尺寸相近等优点,作为一种新型的酶固定化载体在生物技术领域得到了广泛关注[8]。纳米复合材料是由不同性能的纳米材料复合而成,可以有效解决单一纳米材料固定化酶所面临的问题,在保持酶活和增强酶的稳定性等方面有较为突出的表现[9]。本文对近几年在固定化酶领域所采用的纳米复合材料进行了一个系统的分类,并阐述了纳米复合材料的结构和制备过程及其各组分对固定化酶性能产生的影响。
1 硅基纳米复合材料固定化酶研究
由于高比表面积、高化学纯度、高稳定性、化学惰性、无毒和易于修饰等优良特性,以二氧化硅(SiO2)为代表的纳米硅基材料已经成为广泛使用的酶固定化材料[10]。
1.1 磁性硅基纳米复合材料固定化酶
在空气或酸性环境下,磁性纳米粒子(MNPs)的氧化或溶解会大大限制其使用性能,且由于MNP之间存在的磁引力易于聚集而影响分散性能,将MNPs导入硅基纳米材料和将硅基纳米材料覆盖在MNPs表面形成核壳结构是当前广泛使用的保护方法。其中核壳结构纳米复合材料具有核壳结构的结合功能和独特的磁性响应率、低毒性和可化学修饰的表面,在固定化酶领域具有极大的应用潜力,引起了广泛关注[11]。
介孔二氧化硅(mSiO2)作为已知发展最为成熟、研究最为透彻的介孔材料具有独特的孔道结构(2~50nm),不仅有利于大尺寸分子和基团的进入,还因其具有大量纳米级孔道而具有纳米材料的特性。同时,mSiO2材料如MCM-41、MCM-48、SBA-15和介孔泡沫(MCFs)可以增加酶固定化材料的比表面积、提高酶的稳定性和活性,越来越多的研究者将它作为复合材料的基体来制作酶固定化载体[12-13]。
Xie等[14]首先通过化学共沉淀法获得磁性纳米粒子Fe3O4,再通过stober法在其表面形成介孔材料MCM-41,从而获得Fe3O4@MCM-41核壳结构纳米复合材料(图1),然后引入氨基官能团并以戊二醛(GA)为交联剂共价固定褶皱假丝酵母脂肪酶(CRL)。通过透射电子显微镜(TEM)图像分析表明,具有MCM-41外壳的复合载体有效地克服了Fe3O4粒子之间强磁偶极-偶极相互作用。该法的固定化效率为76%,在油脂的Sn-2位酯交换反应中保持优良的催化活性和选择性,催化效率达到228.2U·g-1;重复使用性有所提高,经过5次重复使用酶活基本保持不变,饱和磁化强度值为26.3emu·g-1,可以通过外加磁场达到简易分离。类似的,Zhu等[15]以羧基功能化SiO2包裹的磁性纳米粒子(Fe3O4@SiO2-NH2-COOH)为载体共价固定猪胰脂肪酶(PPL),用于脂肪酶抑制剂的筛选。热重量分析法(TGA)和傅里叶变换红外光谱(FTIR)表征表明,载体成功羧基功能化且通过酰胺键共价固定PPL在其表面;振动样品磁强计(VSM)所得磁滞回线分析表明,载体Fe3O4@ SiO2-NH2-COOH和固定化PPL拥有高饱和磁感应强度,分别达到45.75和42.25emu·g-1,相比未羧基化的Fe3O4@SiO2(51.51emu·g-1),磁感应强度略有下降。固定化PPL与游离酶相比,酶活性、可重用性、热稳定性和储存稳定性都得到明显提升,其动力学参数Km值从游离酶的0.29mmol·L-1变为0.02mmol·L-1,Vmax也从3.16U·mg-1·min-1提高到6.40U·mg-1·min-1,说明固定化后酶与底物的亲和力和催化效率均得到提升。
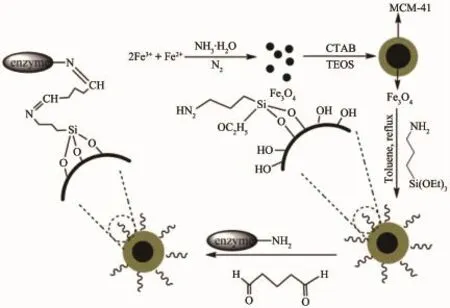
图1 核壳结构磁性固定化脂肪酶的制备过程[14]Fig.1Synthesis of the core-shell structured magnetic immobilized lipase[14]
Kalantari[16]在磁铁矿团簇表面运用溶胶-凝胶法形成无孔SiO2涂层得到载体Fe3O4@SiO2(S1),并利用表面活性剂模板法在Fe3O4@SiO2表面再分别形成介孔(S2)和大孔(S3)2种不同硅基结构的SiO2涂层,从而得到无孔、介孔、大孔3种SiO2涂层磁铁矿团簇纳米复合粒子(图2)。将3种粒子氨基官能化后,以GA为交联剂通过共价结合的方式固定化洋葱假单胞菌脂肪酶(BCL),在外加磁场的情况下都显示出了高稳定性和易于回收的特点。结果表明,硅基涂层结构对于固定化酶的稳定性和催化性能方面有着显著的影响,固定在孔径为1~15nm的介孔硅基材料的酶分子比固定在无孔硅基和大孔硅基结构表现出更高的稳定性和更好的催化性能,而且S2固定化BCL效率(62%)明显高于S1(49%)和S3(58%)。Wu等[17]采用类似的方法得到复合材料Fe3O4@mSiO2共价固定酪氨酸酶,并以无孔SiO2所形成的磁性微球Fe3O4@SiO2做对照组。结果显示Fe3O4@mSiO2具有更高的稳定性和重复使用率,这与Kalantari课题组的研究结果一致。

图2 不同SiO2结构固定化脂肪酶示意图[16]Fig.2Schematic illustration of different silica structures immobilized lipase[16]
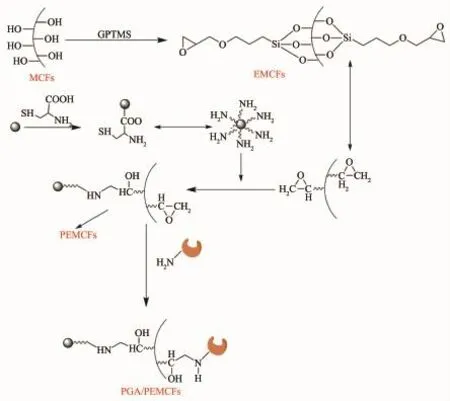
图3 PEMCFs固定化PGA制备过程[18]Fig.3Procedure for immobilization of penicillin G acylase on paramagnetic epoxy-functionalized MCFs[18]

图4 PGA共价固定过程[19]Fig.4Procedure for covalent immobilization of penicillin G acylase[19]
卢冠忠课题组首先通过γ-缩水甘油醚氧丙基三甲氧基硅烷(GPTMS)与MCFs的表面羟基反应得到环氧官能化介孔泡沫(EMCFs),再以L-半胱氨酸氨基官能化Fe3O4纳米粒子表面,将Fe3O4表面氨基与EMCFs外侧环氧基团结合得到顺磁性环氧官能化介孔泡沫(PEMCFs)[18],青霉素酰化酶(PGA)分子通过表面氨基与PEMCFs内侧表面环氧基反应实现共价固定(图3)。固定化PGA具有8800U·g-1的初始活性,且在10次循环后保留94.5%,相比无环氧基的PMCFs固定化PGA稳定性提高了13.6%,相比MCFs固定化PGA稳定性提高了23.3%。在之后的固定化酶研究中,该课题组[19]在中性溶液中以三嵌段共聚物PluronicP123为模板,正硅酸乙酯(TEOS)和三甲氧基硅基丙醛(TMSP)缩聚合成醛基功能化纳米复合材料载体Fe3O4@mSiO2。PGA通过其表面赖氨酸残基的自由氨基与支持材料表面醛基发生希夫碱反应实现共价固定在该顺磁性纳米复合材料(图4);并对制备中TMSP作为硅源的最佳含量进行了研究,结果表明,TMSP在所有硅基组分所占物质的量之比为0.15时固定化酶性能达到最佳,固定化PGA的酶活达到6231U·g-1,虽然与之前研究的MCFs类载体固定化酶方法相比,酶活有所下降,但该法采用一步合成法简易了载体合成步骤,在工业应用中有更广阔的前景;而且固定化酶操作稳定性良好,在10次循环后仍保留91.0%的初始活力,其饱和磁化强度值可达35.1emu·g-1。
1.2 有机聚合物-硅基纳米复合材料固定化酶
功能性聚合物如缩多酸、聚酰胺、聚多元醇、聚酰亚胺和聚醛由于其通用性被广泛应用于新材料的制备,尤其是含有醛基的有机聚合物在酶固定化方面效果显著[20]。
聚乙烯醇(PVA)是一种具有良好生物相容性、生物降解性的水溶性多羟基化合物,PVA复合材料具有良好的机械强度,在生物医学领域得到了广泛应用[21]。PVA可以大幅调节SiO2溶胶-凝胶的内部微环境用于稳定其包埋的酶[22]。Payentko等[23]通过使用溶胶-凝胶法制备SiO2和气相SiO2(A300),将其作为纳米复合材料中2种不同结构特征的硅基组成部分,与由PVA和聚丙烯酸(PAA)组成的聚合物相结合,分别固定化胆碱酯酶(CHE)和乙酰胆碱酯酶(ACHE),催化氯化乙酰胆碱水解反应(图5)。2种纳米复合材料固定化酶均可提高酶催化活性,但在固定化CHE和ACHE方面,以A300为基质的纳米复合材料载体比溶胶-凝胶法制备的SiO2为基质的载体固定化酶的酶活分别要高出19%和5%,并通过扫描电镜(SEM)图像观察,可以得出硅基组分结构的差异在一定程度上影响了固定化酶的催化活性。Singh等[24]采用改良的Stober法合成了PVASiO2纳米杂化材料用于固定α-淀粉酶,固定化酶活性最高可达到21.823U·mg-1,是游离酶初始活性1.25倍,6次循环使用后仍能保留88%的初始活性;固定化酶和游离酶的可溶性淀粉水解动力学研究表明,固定化酶对底物的亲和力远高于游离酶,并且酶的保存限期也得到延长,在35℃储藏25d后保留活性仍高达95.6%,比游离酶高出61%。Shafiee等[25]先利用Stober法合成MCM-41,再通过乳液聚合技术在MCM-41外表面形成聚丙烯醛,从而得到MCM-41@聚丙烯醛核壳纳米复合材料,通过载体表面的醛基与酶的氨基可实现共价固定细毛嗜热霉脂肪酶(TLL)。X射线衍射(XRD)、SEM、TEM等表征结果表明,MCM-41在PVA聚合物表面分散均匀且MCM-41的结构特征得以保留,该纳米复合材料固定化TLL在pH值耐受性、热稳定性、储藏稳定性和重复使用性上展现出优越的性能,在15次循环使用后仍能保留74%的初始活性。

图6 用于制备硅基载体的多糖的化学结构[27]Fig.6Chemical structures of polysaccharides used to prepare silica biocomposites[27]
天然生物材料因其具有独特的性质、来源广泛、环境友好、良好的生物相容性等一系列优点在酶固定化的应用中受到青睐。植酸(PA)是一种天然提取材料,植酸具有很强的螯合能力,分子结构中拥有6个带负电的磷酸根基团,带正电荷的蛋白质与带负电荷的PA之间存在强静电作用,可形成不溶性复合物。Zhao等[26]利用PA与含正电荷的材料或基团之间存在较强相互作用的原理,通过反相微乳法和静电结合法合成PA@SiO2纳米复合材料,用于固定化葡萄糖氧化酶(GOx);圆二色光谱(CD)表明固定化酶仍基本保持其天然二级结构,以PA和硅基材料构建的复合载体具有非常完美的生物活性,可用于构建高灵敏性的生物传感器。De Matteis[27]将右旋糖酐、黄原胶、海藻酸钠和壳聚糖4种多糖分别与硅基载体(图6)相结合,得到一种新型的多聚糖@SiO2复合纳米材料,成功弥补了无机材料在生物相容性方面的不足,并通过溶胶-凝胶法将氯过氧化物酶(CPO)包埋进固定化载体多聚糖@SiO2;通过对照试验发现含有壳聚糖组分的载体固定化酶在各方面展现出了最为优越的性能,且在一定范围内,壳聚糖含量越大,改善固定化酶性能的效果越明显。壳聚糖含量为0.54%时热稳定性和重复使用性达到最高,在高达70℃环境下2h后,仍可以保留超过95%的活性,反应循环次数高达18次。Shahgaldian等[28]设计出一种全新的复合式载体结构用以固定化β-半乳糖苷酶(β-gal):首先通过戊二醛作交联剂以共价结合的方式将β-gal固定在氨基修饰的SiO2纳米粒子的表面;之后通过有机硅烷缩聚反应,3-氨基丙基三乙氧基硅烷(APTES)和正硅酸乙酯(TEOS)在SiO2-(β-gal)表面形成有机硅层,起到对酶的保护作用。相比SiO2-(β-gal),在形成有机硅层后酶活保留68%,但其机械强度、稳定性、重复使用性均得到提升。采用相同的固定化方法对磷酸酶、漆酶、乙醇脱氢酶和天冬氨酸转氨酶进行固定化研究,结果表明通过将有机硅层厚度控制在适当尺寸,将酶包裹在有机硅层内并不会妨碍酶促反应的进行。
1.3 其它硅基复合纳米材料固定化酶
纳米黏土是一种层状铝硅酸盐,因其价格低廉、尺寸小,具有独特的可嵌入属性以及含有大量可作为酶吸附位点的硅醇基团,在固定化酶领域引起了关注[29]。Li[30]通过MNPs有序自组装在纳米粘土特定区域合成超顺磁Fe3O4@Clays纳米复合材料,经过氨基功能化后以GA为交联剂共价固定葡糖淀粉酶(GAS)。通过TEM、FXRD、VSM等表征结果显示Fe3O4@Clays具有高度有序结构、大表面积和高磁敏感性等特性;该复合材料通过在纳米黏土局部位点的修饰降低了由其杂质带来的负面影响;固定化酶的热稳定性和可重用性得到明显提升;其高磁敏性简化了分离过程,更重要的是该材料是一种可再生载体,在固定酶失活后可实现回收再利用,极大降低了在应用中的成本。Ilk等[31]在十八胺-蒙脱石(ODA-MMT)有机黏土表面,通过左旋乳酸(LA)和聚(马来酸酐-甲基乙烯基醚)共聚物(Poly(MA-MVE))的原位共聚作用得到有机-无机纳米复合材料Poly (MA-MVE)-PLA@ODA-MMT,用以固定化漆酶,通过载体结构的设计改善酶所处微环境,从而进一步提高了漆酶催化效率和固定化性能。结果表明,在最优反应条件下,固定化漆酶的催化效率是游离酶的2倍,固定化酶在10次循环使用后仍能保留超过77%的初始酶活,其储藏稳定性也得以进一步提高,可在4℃下保存30d,保留活性是游离酶的3.6倍。
Zhang等[32]通过原位水热法成功地将ZnO纳米线导入mSiO2的孔道,显著增加了mSiO2的比表面积(233m2·g-1),且由于带正电荷的ZnO纳米线(等电点为9.5)与带负电荷的南极假丝酵母脂肪酶CALB (等电点为6.0)之间存在强静电相互作用,这种全新的纳米复合材料对CALB有非常强的吸附作用,酶负载量达到196.8mg·g-1,较单独mSiO2材料提高2~3倍,而且活力达到667.2U·g-1,mSiO2载体固定化酶的5倍,其催化的手性2-辛醇反应在最佳反应条件下,转化率为49.1%,在酶催化手性拆分(R,S)-2-辛醇中对对映体(R)-2-辛醇乙酸酯的选择性高达99%,在15次循环使用后,固定化脂肪酶仍可保留96.9%的相对活性和93.8%的对映体选择性。
银纳米粒子(AgNp)掺入载体材料中可以在保留固定化酶完整催化活性前提下提高其储藏时间,Singh[33]以阿拉伯树胶和明胶为模板采用双模板聚合法聚合四甲氧基硅烷并在其中掺杂AgNp,得到一种新型纳米复合材料。用此材料固定的α-淀粉酶可以在40℃条件下贮藏30d后仍保持其初始活性,而未掺杂AgNp的复合材料在48h会失去31%的活性,相比固定化酶的动力学参数与游离酶相比,Km值为原来的116.4%,Vmax提高了55.1%。
2 碳基纳米复合材料固定化酶研究
碳基材料具有耐高温、导热性能良好、化学惰性、良好的生物相容性等优点,碳基纳米材料如石墨烯、碳纳米管已在固定化酶领域取得了广泛的应用[34-36]。制备碳基纳米复合材料不仅可以保留碳基材料本身所固有的优点,也可克服其韧性差、性能异变、分散性能不好、酶的固定化效率低等缺点。
2.1 石墨烯纳米复合材料固定化酶
石墨烯是一种新型的二维蜂巢晶格结构、单原子厚度的碳基纳米材料,被认为是目前世界上最薄的材料,其在导热率、机械强度、化学稳定性、生物相容性等方面拥有优良的属性,如今被广泛应用于各个领域,在工业应用上具有非常好的前景[37]。氧化石墨烯(GO)是石墨烯的衍生物,非常适合吸附金属氧化物纳米颗粒以及固定大量的酶。GO除了大比表面积和呈现出非常好的生物相容性之外,其表面有环氧基、羧基、羟基等一系列官能团,因此其在水中有很好的分散性并易于化学修饰[38]。在生物领域,石墨烯及其衍生物作为一种与生物分子有强相互作用的新兴纳米材料已被运用于生物传感器的制备,如Li等[39-40]利用石墨烯量子点修饰的热解石墨烯电极[39]及GO修饰金电极[40]来获得灵敏度更高、选择性更强的电化学传感器。
2.1.1 磁性石墨烯纳米复合材料固定化酶
GO作为固定化材料由于其强亲水性很难从水溶液中分离,并且GO纳米薄片之间由于存在强ππ键相互作用容易结块,从而导致大量官能团被覆盖[41-42]。为了进一步拓展和优化GO在生物技术上的应用,将其与磁性纳米粒子结合形成复合材料,可以有效地改善其性能。
Zhang等[43]通过溶剂热法制备功能化Fe3O4磁性纳米粒子,其氨基和化学沉淀法制备的GO的羧酸基团反应,共价连接形成磁性纳米复合材料GOCO@NH-Fe3O4(图7),该材料再以π-π堆积作用和氢键相互作用固定化胰蛋白酶(TPS),固定化酶的比重达到0.275mg·mg-1,其稳定性也有所提升,可在4℃条件下储藏30d后仍可保留84%的初始活性。GO-CO@NH-Fe3O4复合材料除了GO和Fe3O4本身所带有的特性,还具有对微波辐射良好的吸收性能,在实际应用中可通过微波辅助进一步提高固定化TPS对蛋白质的消化效率,分解效率是无微波辅助的传统酶溶液分解法的2880倍。Jiang等[41]利用超声波辅助共同沉淀法成功地将溶液中的FeSO4·7H2O和FeCl3·6H2O在GO片表面形成Fe3O4纳米粒子,并用于固定辣根过氧化物酶(HRP)。固定化酶的生化性质都得到一定的改善:贮藏稳定性、PH值和温度的耐受性相比游离酶都得到加强;重复使用性得到了显著增强,固定化HRP在4次循环后仍保留66%的初始活性。Chen[42]同样采用Fe3O4@GO复合材料来固定化乙醇脱氢酶(ADH),除了对pH值耐受性和热稳定性得到加强,重复使用性也得以提高,10次使用后,仅失去大约20.4%的活力。GO制备过程中在一定程度上会破坏石墨烯表面规整度,通过化学还原法制备还原氧化石墨烯(rGO)可以在保留GO分散性的同时恢复其固有的优良性能。Shi等[44]以GO为氧化剂氧化Fe2+以得到Fe3O4纳米粒子,粒子沉积在还原氧化石墨烯(rGO)表面形成纳米复合材料rGO@Fe3O4,此材料同时结合了Fe3O4纳米粒子的磁性能和rGO纳米片的大比表面积的特点,过氧化氢酶(CAT)通过氢键和疏水相互作用直接吸附在rGO@Fe3O4纳米复合材料表面。CAT负载量高达(312.5±12.6)mg·g-1,固定化酶活性回收率达到98%,除了具有很强的磁响应外,在稳定性和可重用性上表现出优良的性能。
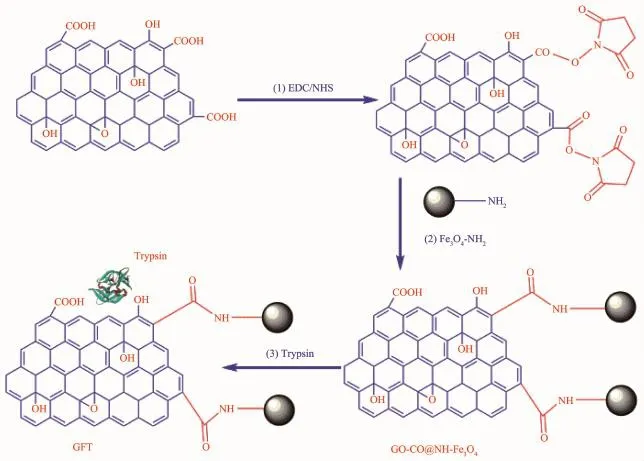
图7 GO-CO@NH-Fe3O4的制备过程[43]Fig.7Procedure for GO-CO@NH-Fe3O4fabrication[43]
随着酶负载量的增加,酶分子间形成空间位阻从而限制底物和产物之间的扩散,影响酶催化活性,研究固定化过程中酶的添加量也具有重要意义。Wu等[45]采用水热合成法合成Fe3O4@rGO纳米复合材料用以固定化HRP,用以进一步研究载体HRP负载量和HRP初始加入量之间的数量关系,以及负载量增加对固定化HRP保留活性的影响。结果表明,HRP负载量和HRP初始加入量呈线性关系,在HRP加入量为25mg·g-1时达到稳定;HRP负载量很低时,固定化HRP可以保持90%的高活性,但其会随着负载量的增大而降低直至达到平衡状态。该载体最佳HRP负载量为23.3mg·g-1,固定化效率高达91%,在10次循环使用后仍保留70%的初始活性。Jing等[46]将已制备好的Fe3O4@MCM-41结合到GO表面,并以3-氯丙基三乙氧基硅烷(CPS)修饰GO表面形成新型纳米复合材料CPS/GOFe3O4@MCM-41,用来共价固定PPL(图8)。Fe3O4纳米粒子不仅可以通过外加磁场实现快速分离,更重要的是防止GO的集聚从而使材料表面区域和功能基团得以保留。实验结果表明该复合材料的固定化效率和最大相对活性分别达到98%和97.9%。固定化酶在30℃下保存56d仍可保留85%的活性,远高于游离酶的19%,10次循环使用后的保留活性仍高达87%,表明该GO复合材料在提高稳定性和可重用性方面具有重要作用。值得一提的是,该法中酶和支持材料之间几乎不存在空间位阻,这大大提高了催化效率。
2.1.2 有机聚合物-石墨烯纳米复合材料固定化酶
聚合离子液体(PILs)兼具离子液体和聚合物两者优点,如良好的电导性、热稳定性和机械强度等,PILs在合成各种纳米材料中是非常有效的稳定剂或改性剂。近年来,利用聚合离子液体修饰石墨烯表面改善其在水溶剂中的可分散性和溶解性成为一种行之有效的方法[47]。Li等[48]首先用NaBH4预处理GO表面得到rGO,再通过原位聚合法,通过合成聚合离子液体1-乙烯基-3-丁基咪唑溴化物(ViBuIm+Br-)形成纳米复合材料poly(ViBuIm+Br-)@ GO,通过静电作用将GOx固定在其表面,poly (ViBuIm+Br-)不仅改善了载体的疏水性,且在水溶液中可提供正电荷以进一步在温和条件下实现酶的固定化。

图8 CPS/GO-Fe3O4@MCM-41-PPL的制备过程[46]Fig.8Procedure for GO-CO@NH-Fe3O4fabrication[46]
壳聚糖(CS)是一种天然多糖,具有多种官能团,无毒性,拥有良好的吸附性能、生物相容性和生物降解能力,但其机械强度差的缺点一直限制其应用。将GO和CS通过共价连接制备GO@CS复合材料可以较好地解决上述缺点。Li等[49]首先通过Hummers法制备GO,再使用EDC/NHS为缩合剂将CS共价连接到GO,FeCl3·6H2O和1,6-己二胺通过溶剂热反应在其表面生成Fe3O4纳米粒子从而得到复合材料CS-GO@Fe3O4(图9),并通过载体静电吸附(Ⅰ法)、以GA做交联剂共价结合(Ⅱ法)、以亚氨基二乙酸(IDA)和Cu2+修饰载体通过金属螯合作用(Ⅲ法)固定化CAL,3种固定化方法都提高了CAL对pH值和温度的耐受性,最适温度范围从游离酶的45~50℃,拓宽到45~65℃,且在75℃下仍可保留75%的活性;Ⅱ法在重复使用性上表现最为优异,且10次循环使用后仍可保留70%的保留活性。
2.2 碳管纳米复合材料固定化酶
碳纳米管作为一种一维纳米材料具有良好的导电性、吸附能力强、生物相容性好等许多独特属性,随着对碳纳米管及纳米材料研究的不断深入,碳纳米管在生物技术领域也被广泛使用[50-51]。
Li等[52]通过Cu2+螯合作用将GAS固定在表面接有聚酰胺-胺型树枝状高分子(PAMAM)的磁性碳纳米管上,考察了该方法固定化GAS的相关性能。结果表明酶的负载量达20.6mg·g-1,固定化酶的活性为游离酶初始活性的71.2%,经过10次循环使用后,固定化酶仍能保持84.2%初始活性,且载体再生后仍然保留着其原有优良的磁分离能力、快速固定化酶能力等特性(图10)。Choi[53]将多壁碳纳米管(CNT)在全氟磺酸(Nafion)溶液中分散氧化形成CNT@Nafion纳米复合材料,并通过等离子腐蚀在其表面获得的微孔结构共价固定GOx和漆酶,其固定化效率得到明显改善。Xu等[54]制成1-芘丁酸改性掺氮碳纳米管(PBA@NCNTs)均质纳米复合材料,用于酶的固定化和葡萄糖生物传感的研究,GOx在PBA/NCNTs表面酶负载量高达1.986mmol·cm-2。而Amatatongchai[55]将氨基功能化碳纳米管(CNTNH2)、金纳米粒子和牛血清白蛋白组成纳米复合材料用以共价固定漆酶,并考察了该酶电极上漆酶直接电化学和生物传感性能,结果表明合成的纳米复合材料有良好的酶直接电化学活性,也保持了漆酶的生物活性,同时该生物传感器具有良好的选择性、重复使用性及稳定性。
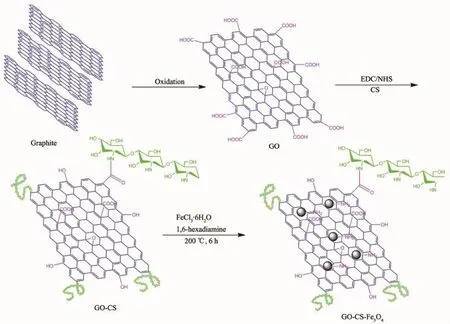
图9 CS-GO@Fe3O4合成过程示意图[49]Fig.9Schematic illustration of the synthesis process used to produce CS-GO@
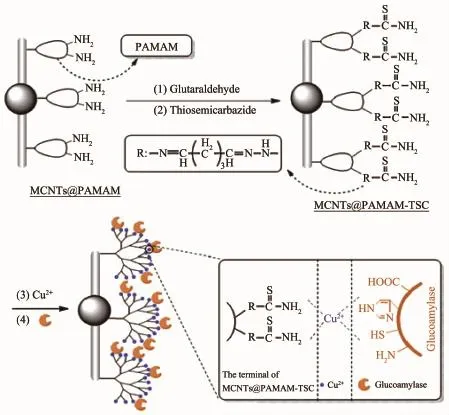
图10 MCNTs@PAMAM-TSC固定化GAS过程示意图[52]Fig.10Schematic illustration of GAS immobilized onto MCNTs@PAMAM-TSC[52]
3 纳米纤维复合材料固定化酶研究
3.1 聚合物纳米纤维复合材料固定化酶
纳米纤维复合材料固定化酶易从反应介质中分离并可应用于连续操作,因此被广泛应用于催化、生物医学和制药领域[56]。
静电纺丝是一种将各种聚合物、高分子共混物、溶胶-凝胶、复合材料等制备成纤维直径在1~100nm的多功能纳米纤维的技术,由于其制造工艺简单、成本低廉等优点成为世界材料科学技术领域研究的热点[57]。El-Aassar[58]通过静电纺丝技术制得聚甲基丙烯酸甲酯-丙烯腈(poly AN-co-MMA)纳米纤维,并以纳米纤维羰基基团共价连接的聚乙烯亚胺(PEI)为连接臂共价固定化β-半乳糖苷酶。研究表明,耦合PEI数量是影响固定化酶催化性能、保留活性和动力学参数的主要因素。检测结果表明,固定化酶的稳定性得到明显的改善,最适温度比游离酶高5℃且对温度具有更好的耐受性。
Manuel[59]通过相同的技术制得聚丙烯腈(PAN)纳米纤维薄膜,并通过2种不同的表面化学修饰后作为固定化葡糖氧化酶的载体。一是通过两步法将PAN的腈基转化为氨基;二是先通过NaOH处理生成羧酸基团,再进一步与己二胺(HMDA)反应生成反应性间隔臂来固定化酶。结果表明,化学修饰后载体表面没有发生变化且其机械强度得以改善,一法和二法修饰所得载体的固定化酶在25次循环后分别保留54%和60%的初始活性,酶负载量分别为1238μg·mg-1和168μg·mg-1;且保留活性和动力学参数方面都展现出更为优良的性能。
3.2 纤维素纳米纤维复合材料固定化酶
纤维素是自然界分布最广、含量最多的一种天然高分子,具有可降解、可再生、密度小以及对生态环境不产生污染等优点。但是,天然纤维素不管在物理形态还是化学性能上都存在一些缺陷,例如溶解性差、不耐化学腐蚀、强度有限等。纳米微晶纤维素(NCC)是指在尺度上至少有一维达到100nm或100nm以下的纤维素,同纤维素或微晶纤维素相比,NCC不仅具备生物相容性、更高的拉伸强度、更多的结晶区和更高的结晶度,而且在纳米尺度下,其表面暴露着大量羟基,使得NCC具有高亲水性、高稳定性与高反应性,同时还具有高透明性与液晶性以及比表面积高的特点。NCC在酶固定化、药物传输、生物医学等方面都得到了广泛的应用[60-61]。NCC与纳米材料复合可以得到性能优越的复合载体用于固定化酶,且可以很好地提高其机械强度。Xu等[62]通过在CS/PVA纳米纤维薄膜中添加NCC以解决原本机械强度差的问题。结果显示,加入5% NCC的纳米复合薄膜的拉伸强度比未加入的高出3.7倍;固定化HRP的最高酶载量达到384mg·g-1,CS/PVA-NCC薄膜相比CS/PVA薄膜固定化酶的稳定性和可重用性进一步提高,去除3,3′,5,5′-四溴双酚(TBBPA)效率从14.6%·h-1提升至32.0%·h-1,在工业处理废水上具有潜在的应用。尽管NCC具有很多优异性能,但由于其在水中良好的分散性,NCC很难从反应体系中分离,从而限制了在工业上的应用,而利用NCC制备磁性纳米复合材料是一个很好的选择。NCC和Fe3O4表面都带负电荷,因此Fe3O4很难稳定吸附在NCC表面,而CS是生物相容性良好的天然亲水性阳离子多糖。娄文勇[63]课题组利用CS和NCC、Fe3O4之间存在的静电相互作用,采用共沉淀-静电自组装技术合成磁性纤维素纳米晶体复合物MNCCs(图11),用以共价固定化木瓜蛋白酶(PAP),成功解决了NCC材料不易回收的问题。固定化后的PAP呈现出更高的热稳定性,在40℃下存储7h后仍能保持高于80%的相对活性,而游离酶则低于30%;同样pH值稳定性也有所提高,适应范围从5~7提高到5~10;储藏性能十分优越,可在40℃储藏16d后仍保持93.6%的相对活性;且固定化后的催化效率显著优于游离酶。
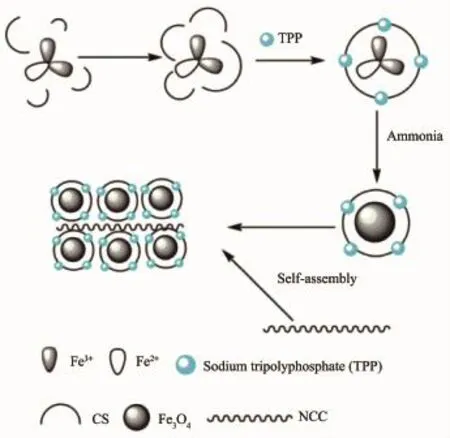
图11 MNCCs的合成路线[63]Fig.11Synthetic route of MNCCs[63]
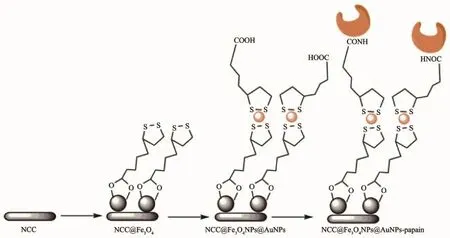
图12 NCC@Fe3O4NPs@AuNPs制备及固定化酶示意图[65]Fig.12Schematic illustration for the preparation and immobilized enzymes[65]
后来该课题组[64]又采用共同沉淀-交联技术,先使Fe3O4与NCC静电结合,再以CS覆盖其表面合成MNCCs,并用于固定化PAP来进行二肽丙氨酰谷氨酰胺(Ala-Gln)的高效生物合成,MNCCs的酶负载量达到333mg·g-1,保留活性达到80%。Mahmoud[65]同样采用磁性纳米微晶纤维素作为固定化PAP的载体(图12),与娄文勇课题组不同的是,Mahmoud将Fe3O4和Au两种纳米粒子固定在NCC表面。AuNPs易于制备,具有可靠的化学稳定性、生物相容性、通用性,是一种可用于修饰Fe3O4NPs表面从而达到保护Fe3O4目的的理想材料;NCC@Fe3O4NPs@AuNPs纳米复合材料最优酶负载量为186mg·g-1,且在4℃下储藏35d后可保留95%的初始活性。
4 其它纳米复合材料固定化酶研究
固定不同种类的酶或在不同的反应体系中,对固定化酶的要求也会有相应的变化,根据实际情况来决定复合材料种类已成为制备纳米复合载体的常用方法。
四氧二铁酸钴(CoFe2O4)磁性纳米粒子是一种有卓越的生物相容性,易于分离的材料,其具有高结合能力和优良的磁性、化学稳定性和机械强度。Altun[66]用水热法合成CoFe2O4磁性纳米粒子,再以鞣酸(TA)覆盖在其表面得到复合材料CFO-TA,用于GOx的固定化(图13)。因蛋白质和TA之间存在强相互作用,GOx可以通过简单的物理吸附达到很好的固定化效果。与其它固定化方法相比,此法不需要各种化学反应和复杂的过程,简单易行、耗时短,且不会对酶结构造成破坏。根据双倒数分析(LB)法可得固定化GOx对葡萄糖拥有低亲和力,固定化酶和游离酶的Km常数为50.05和28.00mmol·L-1,固定化酶重复使用性得以提升,在8次连续使用后仍可保留60%的初始活性。
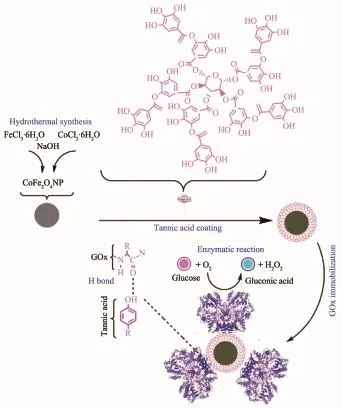
图13 CFO-TA固定化GOx及其酶促反应示意图[66]Fig.13Schematic illustration of GOx immobilization onto CFO and GOx enzymatic action[66]
Gogoi等[67]通过等离子体聚合和溅射技术将AgNPs均匀地嵌入苯胺聚合薄膜中得到Ag/PPAni纳米复合薄膜。AgNPs的加入可以较好地保留酶的活性,而PPAni作为一种在环境中稳定的聚合物,其表面存在大量具有可供固定化酶结合的活性位点的自由基,因此Ag/PPAni不需要借助任何表面活性剂或GA等交联剂即可与酶稳定共价结合。PPAni具有的生物相容性和高抗微生物活性也是选择其做固定化载体关键的因素,Ag/PPAni纳米复合薄膜所固定的胰蛋白酶的水解能力远高于游离酶,在蛋白质消化领域用作理想载体有非常好的应用前景。Kim等[68]通过逐步聚合法将Fe3O4嵌入聚苯乙烯/聚吩的核/壳复合物粒子中形成多功能Fe3O4NPs-PSt/PTh纳米复合材料(图14),此法获得产物在导电性能上表现优异,多用于生物酶传感器的制备。
羟磷灰石(HA)作为一种具有低成本、良好生物降解性和生物相容性等优良性能的无机生物材料在固定化酶领域得以应用。Faramarzi等[69]首先以Fe3O4纳米粒子为内核,以HA作为表面涂层合成Fe3O4NPs@HA,再在外层通过包裹具有大量氨基官能团的聚乙烯亚胺(PEI)加以保护,最后通过表面嫁接可以有效保护酶空间结构的β-环糊精(β-CD)得到3层核壳结构的纳米复合材料Fe3O4NPs@HA @PEI-β-CD,此法获得的载体固定化脂肪酶在pH值耐受性,热稳定性和储藏稳定性等方面有了明显的提升,其在经过5次循环使用后可保留80%的初始活性。
纳米CaCO3成本低廉易于合成,具有高机械强度和热稳定性,并且可以为固定化酶提供高亲水微环境,但由于其表面官能团密度非常低,因此表面吸附能力较低。Preety[70]通过细乳液法制备CaCO3纳米粒子,再将环氧氯丙烷(环氧树脂)和丙二酚(硬化剂)聚合在聚乙烯层表面得到具有环氧基团的聚乙烯层,将其与CaCO3纳米粒子结合得到Epoxy@ CaCO3纳米复合载体。由于环氧基团和酶之间存在多种共价连接,可以成功解决上述问题,并且与纳米CaCO3的结合降低了环氧基材料对酶活造成的损失并提高酶的负载量。Epoxy@CaCO3纳米复合载体成功地固定化CAT,其酶负载量和保留活性分别为0.67mg·cm-2和92.63%,比未结合CaCO3纳米粒子的Epoxy载体分别提升了24%和35%;且CAT的热稳定性和储藏稳定性得到明显提升,在75℃下1h固定化酶保留活性是游离酶的3倍,在5℃磷酸盐缓冲液中储藏,固定化酶半衰期比之前延长了5倍;且在重复使用30次后酶初始活性未有明显损失。
Zhai等[71]通过将CS组装在埃洛石纳米管(HNTs)表面合成出CS-HNTs(图15)。通过N2吸附-脱附、FTIR、TEM、SEM等表征分析,发现其结构为层状多孔结构。该载体在共价固定HRP上展现了优越的性能,最大酶负载量达到21.5mg·g-1,远高于未复合的埃洛石固定的3.1mg·g-1。经过35d的储藏,固定化HRP没有损失任何活力,而游离酶仅保存其27%的初始活力。Krishnan等[72]利用金属纳米粒子有助于增强复合材料的电催化、光催化等功能的特性,通过AgNp与APTES修饰过的HNTs表面氨基基团的配位作用得到AgNp@HNT纳米复合材料,其酶负载能力达到进一步提升,可达到168mg·g-1;储藏稳定性也得以加强,在30d后仍可保留超过90%的初始活性;且由于金属离子的加入,AgNp@HNT也展现出优越的电子导电率、电催化活性等电化学性能,在生物传感器方面也有较好的应用前景。
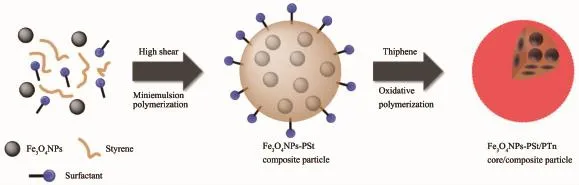
图14 Fe3O4NPs-PSt/PTh示意图[68]Fig.14General scheme of Fe3O4NPs-PSt/PTh[68]
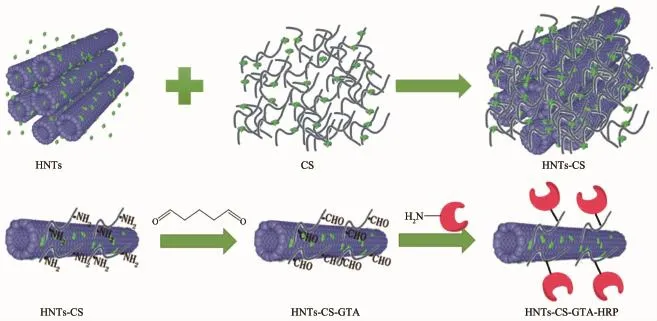
图15 CS-HNTs的制备和固定化机制示意图[71]Fig.15Schematic illustration of preparation and immobilization mechanism for CS-HNTs[71]
5 总结与展望
随着纳米材料研究的不断深入,纳米复合材料在固定化酶领域已经广为研究,并取得大量成果,尤其在硅基材料和碳基材料上研究得最为广泛。利用纳米材料得良好生物相容性、大比表面积及易于修饰的表面等特性,可以提高酶负载量和保留最大酶活,如介孔硅和石墨烯已成为实验室固定化酶的常用载体。将拥有不同特性的纳米材料复合得到新的载体,可以在继承其本身所固有优点的同时解决单一材料某些方面的缺点,如固定化效率低、分离困难、影响酶结构等。
尽管如此,纳米复合材料固定化酶仍存在一些需要进一步研究、解决的问题:(1)复合的纳米材料在制备上与单一材料固定化酶相比,流程较为复杂,成本较为昂贵,限制了纳米复合材料固定化酶在工业上的应用;(2)通常纳米复合材料多需要进行表面修饰,一些较为复杂的表面修饰方法会在一定程度上降低酶的活性,利用复合材料本身对酶强大的吸附能力实现固定化方面的研究较少;(3)复合材料种类的选择具有一定的盲目性,大多是根据材料原本属性进行尝试得到相应复合载体,未能从酶结构特征和实际应用出发设计高效的固定化载体,所得载体未必是复合性能最佳的选择;(4)现阶段研究的重点主要放在纳米复合材料固定化酶的制备过程上,对于所得固定化酶的催化反应应用方面的研究较少,限制了纳米复合材料进一步在工业上的应用。
发展制备简易、更为高效、适用性广的纳米复合材料仍需要我们进一步深入研究。通过分子模拟和表征分析,研究载体和酶之间的结合机理和酶活性部位构象的变化机制,从而理性设计固定化载体材料种类及结构,进一步解决固定化载体所带来的分配效应、空间障碍效应和扩散限制效应,深入探讨酶与载体表面之间的作用机理,可以更深更广的拓展纳米复合材料固定化酶的应用范围,将使其在生物传感器、生物燃料电池、污水处理、药物研制等方面拥有光明的发展前景。
[1]Tran D N,Balkus K J.ACS Catal.,2011,1(8):956-968
[2]Rodrigues R C,Ortiz C,Berenguer-Murcia A,et al.Chem. Soc.Rev.,2013,42(15):6290-6307
[3]Zhang Y,Ge J,Liu Z.ACS Catal.,2015,5(8):4503-4513
[4]Dicosimo R,Mcauliffe J,Poulose A J,et al.Chem.Soc.Rev., 2013,42(15):6437-6474
[5]Adlercreutz P.Chem.Soc.Rev.,2013,42(15):6406-6436
[6]Sheldon R A,van Pelt S.Chem.Soc.Rev.,2013,42(15):6223-6235
[7]Min K,Yoo Y J.Biotechnol.Bioprocess Eng.,2014,19(4):553-567
[8]Ansari S A,Husain Q.Biotechnol.Adv.,2012,30(3):512-523
[9]Verma M L,Puri M,Barrow C J.Crit.Rev.Biotechnol.,2016,36(1):108-119
[10]Li Z,Barnes J C,Bosoy A,et al.Chem.Soc.Rev.,2012,41(7):2590-2605
[11]Liu J,Qiao S Z,Hu Q H,et al.Small,2011,7(4):425-443
[12]Yang P,Gai S,Lin J.Chem.Soc.Rev.,2012,41(9):3679-3698
[13]Wu S,Mou C,Lin H.Chem.Soc.Rev.,2013,42(9):3862-3875
[14]Xie W,Zang X.Food Chem.,2016,194:1283-1292
[15]Zhu Y,Ren X,Liu Y,et al.Mater.Sci.Eng.C,2014,38: 278-285
[16]Kalantari M,Kazemeini M,Arpanaei A.Biochem.Eng.J., 2013,79:267-273
[17]Wu S,Wang H,Tao S,et al.Anal.Chim.Acta,2011,686(1/ 2):81-86
[18]Yang L,Gao Z,Guo Y,et al.Microporous Mesoporous Mater., 2014,190:17-25
[19]Yang L,Guo Y,Zhan W,et al.Microporous Mesoporous Mater.,2014,197:1-7
[20]Fang Y,Huang X,Chen P,et al.BMB Rep.,2011,44(2):87-95
[21]Peng Z,Kong L X,Li S D.Synth.Met.,2005,152(1/2/3SI1): 25-28
[22]Keeling-Tucker T,Brennan J D.Chem.Mater.,2001,13(10): 3331-3350
[23]Payentko V,Matkovsky A,Matrunchik Y.Nanoscale Res. Lett.,2015,10(82):1-3
[24]Singh V,Singh D.Process Biochem.,2013,48(1):96-102
[25]Motevalizadeh S F,Khoobi M,Shabanian M,et al.Mater. Chem.Phys.,2013,143(1):76-84
[26]Zhao W,Ni Y,Zhu Q,et al.Biosens.Bioelectron.,2013,44: 1-5
[27]De Matteis L,Germani R,Mancini M V,et al.Appl.Catal. A,2015,492:23-30
[28]Correro M R,Moridi N,Schutzinger H,et al.Angew.Chem. Int.Ed.,2016,55(21):6285-6289
[29]Tzialla A A,Pavlidis I V,Felicissimo M P,et al.Bioresour. Technol.,2010,101(6):1587-1594
[30]Zhao G,Wang J,Li Y,et al.J.Phys.Chem.C,2011,115(14):6350-6359
[31]Ilk S,Demircan D,Saglam S,et al.Turk.J.Chem.,2016,40(2):262-276
[32]Shang C,Li W,Zhang R.Enzyme Microb.Technol.,2014, 61-62:28-34
[33]Singh V,Ahmed S.Int.J.Biol.Macromol.,2012,50(2):353-361
[34]Singh V,Joung D,Zhai L,et al.Prog.Mater.Sci.,2011,56(8):1178-1271
[35]Balandin A A.Nat.Mater.,2011,10(8):569-581
[36]Wan X,Zhang C,Yu D,et al.Prog.Chem.,2015,27(9):1251-1259
[37]Huang X,Qi X,Boey F,et al.Chem.Soc.Rev.,2012,41(2): 666-686
[38]Zhu Y,Murali S,Cai W,et al.Adv.Mater.,2010,22(46): 5226
[39]Zhao J,Chen G,Zhu L,et al.Electrochem.Commun.,2011, 13(1):31-33
[40]Li X,Miao P,Ning L,et al.Curr.Nanosci.,2014,10(6):801-806
[41]Chang Q,Jiang G,Tang H,et al.Chin.J.Catal.,2015,36(7):961-968
[42]Liu L,Yu J,Chen X.J.Nanosci.Nanotechnol.,2015,15(2): 1213-1220
[43]Jiang B,Yang K,Zhao Q,et al.J.Chromatogr.A,2012,1254: 8-13
[44]Yang D,Wang X,Shi J,et al.Biochem.Eng.J.,2016,105(A):273-280
[45]Wu X,Zhang Y,Wu C,et al.Trans.Nonferrous Met.Soc. China,2012,22(S1):S162-S168
[46]Shao Y,Jing T,Tian J,et al.RSC Adv.,2015,5(126):103943-103955
[47]Shen J,Hu Y,Li C,et al.Small,2009,5(1):82-85
[48]Zhang Q,Wu S,Zhang L,et al.Biosens.Bioelectron.,2011, 26(5):2632-2637
[49]Wang J,Zhao G,Jing L,et al.Biochem.Eng.J.,2015,98: 75-83
[50]De Volder M F L,Tawfick S H,Baughman R H,et al. Science,2013,339(6119):535-539
[51]Mehra N K,Mishra V,Jain N K.Biomaterials,2014,35(4): 1267-1283
[52]Zhao G,Li Y,Wang J,et al.Appl.Microbiol.Biotechnol., 2011,91(3):591-601
[53]Choi S D,Choi J H,Kim Y H,et al.Microelectron.Eng., 2015,141:193-197
[54]Xu X,Yu J,Qian J,et al.IEEE Sens.J.,2014,14(7):2341-2346
[55]Amatatongchai M,Sroysee W,Laosing S,et al.Int.J.Electrochem.Sci.,2013,8(8):10526-10539
[56]Sulaiman S,Mokhtar M N,Naim M N,et al.Appl.Biochem. Biotechnol.,2015,175(4):1817-1842
[57]Greiner A,Wendorff J H.Angew.Chem.Int.Et.,2007,46(30):5670-5703
[58]El-Aassar M R,Al-Deyab S S,Kenawy E.J.Appl.Polym. Sci.,2013,127(3):1873-1884
[59]Manuel J,Kim M,Dharela R,et al.J.Biomed.Nanotechnol., 2015,11(1):143-149
[60]Mahmoud K A,Mena J A,Male K B,et al.ACS Appl.Mater. Interfaces,2010,2(10):2924-2932
[61]Klemm D,Kramer F,Moritz S,et al.Angew.Chem.Int.Ed., 2011,50(24):5438-5466
[62]Xu R,Tang R,Liu S,et al.RSC Adv.,2015,5(79):64091-64097
[63]Cao S,Li X,Lou W,et al.J.Mater.Chem.B,2014,2(34): 5522-5530
[64]Cao S,Xu H,Li X,et al.ACS Sustainable Chem.Eng., 2015,3(7):1589-1599
[65]Mahmoud K A,Lam E,Hrapovic S,et al.ACS Appl.Mater. Interfaces,2013,5(11):4978-4985
[66]Altun S,Cakiroglu B,Ozacar M,et al.Colloids Surf.B, 2015,136:963-970
[67]Gogoi D,Barman T,Choudhury B,et al.Mater.Sci.Eng.C, 2014,43:237-242
[68]Kim Y S,Lee S M,Govindaiah P,et al.Synth.Met.,2013, 175:56-61
[69]Khoobi M,Khalilvand-Sedagheh M,Ramazani A,et al.J. Chem.Technol.Biotechol.,2016,91(2):375-384
[70]Preety,Hooda V.Appl.Biochem.Biotechnol.,2014,172(1): 115-130
[71]Zhai R,Zhang B,Wan Y,et al.Chem.Eng.J.,2013,214: 304-309
[72]Kumar-Krishnan S,Hernandez-Rangel A,Pal U,et al.J. Mater.Chem.B,2016,4(15):2553-2560
Research Progress on Enzyme Immobilized on Nanocomposites
XIANG Xin-RanHUANG HeHU Yi*
(State Key Laboratory of Materials-Oriented Chemical Engineering,The Synergetic Innovation Center for Advanced Materials, School Of Pharmaceutical Sciences,Nanjing Tech University,Nanjing 210009,China)
The choice of carrier material has a crucial influence on the performance of the immobilized enzyme. Nanocomposites,which not only have the properties of nanoscale,but also overcome the shortcoming of a single material,have attracted tremendous attention in the field of immobilized enzyme.In this paper,classifications of nanocomposite carriers which are currently used in the field of immobilized enzyme are systematically elaborated; the preparation and the significantly enhanced enzymology properties of enzymes immobilized on Si-based nanocomposites,C-based nanocomposites and nanofibers composites are introduced.The outlook of enzymes immobilized on these nanocomposites is also prospected.
nanocomposites;immobilized enzyme;Si-based nanomaterials;C-based nanomaterials
O613.7;Q81;TB33
A
1001-4861(2017)01-0001-15
10.11862/CJIC.2017.016
2016-07-26。收修改稿日期:2016-11-07。
国家杰出青年科学基金(No.21225626)、国家自然科学基金(No.21676143)和江苏高校青蓝工程资助项目。
*通信联系人。E-mail:huyi@njtech.edu.cn
