掺杂单层MoS2电子结构的第一性原理计算
2016-12-22伏春平
伏春平
(1 重庆文理学院 电子信息技术与应用工程中心,重庆402160;2 重庆市高校微纳米材料工程与技术重点实验室,重庆 402160)
掺杂单层MoS2电子结构的第一性原理计算
伏春平1,2
(1 重庆文理学院 电子信息技术与应用工程中心,重庆402160;2 重庆市高校微纳米材料工程与技术重点实验室,重庆 402160)
采用第一性原理研究Cu, Ag, Au掺杂单层MoS2的键长畸变、能带结构和态密度。探讨Cu, Ag, Au掺杂对单层MoS2电子结构的影响。结果表明:Cu, Ag, Au在S位掺杂的杂质能都低于在Mo位掺杂的杂质能,其在S位掺杂的体系的稳定性强于在Mo位掺杂的体系。在S位掺杂时,杂质与最近邻的Mo,S原子的键长都发生了畸变,畸变率最大的是dAu-Mo,达23.8%。与单层 MoS2的超胞相比,掺杂体系的禁带中出现了4条新能级,导带和价带的能量向低能区移动。杂质原子周围存在着电荷聚集,同时也存在电荷损失。
MoS2;能带结构;态密度;掺杂;第一性原理
MoS2最常见的低维结构为二维单层结构,也是MoS2三维母体材料的基本组成单元。其结构特征类似石墨烯,属于典型的二维原子晶体,拥有二维原子晶体各向异性的结构特点,同时继承了MoS2母体的优异性质,单层MoS2具有典型的层状结构特点,由紧密结合的“夹心面包式”S-Mo-S三个原子层组成,上下两个为硫原子,而中间的原子层则为Mo,三个原子层中的原子都按类似石墨烯的平面六角阵列方式排列,各个分子层的层厚为0.65nm。 层内原子以共价键结合, 层间原子则主要由范德华力结合[1-4]。单层MoS2的禁带宽度为1.8eV ,是直接带隙半导体; MoS2具有众多优异的性质,如催化、能量存储、润滑以及光电性质,在传感器、光电子器件、异质结构器件、场效应管、自旋电子器件等方面具有潜在的应用价值[5-15]。近来,张昌华等[7]的研究表明,Te掺杂单层MoS2间接带隙半导体材料,使单层MoS2的静态介电常数增大,禁带宽度变窄,吸收光谱产生红移;而吴木生等[8]用Cr,W掺杂单层MoS2发现,W掺杂对能带结构几乎没有影响, 但Cr掺杂则影响很大。曹娟等[9]研究了过渡金属V, Cr, Mn掺杂单层MoS2、电子结构、磁性和稳定性;雷天民等[10]研究了稀土元素La, Ce,Nd掺杂单层MoS2,能带结构表明La掺杂可以在MoS2的禁带中引入受主能级, Ce,Nd掺杂可能形成施主、受主能级共存的情况。本工作通过同样的手段来调控单层MoS2结构和电学特性,在第一性原理计算的基础上,研究了Cu, Ag, Au在Mo位和S位掺杂单层MoS2的结构和性能,并系统计算了能带结构和态密度。
1 计算模型和方法
基于密度泛函理论的第一性原理数值基组的方法,采用量子力学程序dmol3完成计算。计算中, 交换关联能为广义梯度近似(General Gradient Approximation, GGA)的Perdew-Burke-Ernzerhof(PBE)泛函[13],倒空间k格点的k-point设置为5×5×1积分网格对布里渊区积分。 优化标准为原子间作用力≤0.001eV/nm, 原子的最大位移≤5.0×10-5nm, 晶体的内应力≤0.02GPa,能量收敛精度≤5.0×10-6eV/atom。本工作采用单层MoS2,5×5×1的超晶胞模型,由75个原子构成;是通过沿MoS2单胞基矢方向分别扩展5个单位得到。在MoS2中,参与计算的分别是Mo的[Kr]4d55s1和S的[Ne]3s23p4电子组态,而Cu, Ag, Au的电子组态分别为[Ar]3d104s1,[Kr]4d105s1,[Xe]4f145d106s1,考虑到单层MoS2中存在Mo位和S位掺杂的不同,分别用Cu, Ag, Au原子替换单层MoS2中一个Mo或S原子来实现替位式掺杂研究。 同时,为了避免层间作用,将层间真空层设为1.8nm,所建模型的俯视图及侧视图如图1所示。计算单层MoS2的本征结构的能带结构,得到本征MoS2的带隙宽度为1.792eV,导带底和价带顶都位于Q点, 是直接带隙能带结构, 计算得到的结果与文献[10]符合,说明选用的研究方法科学可靠。
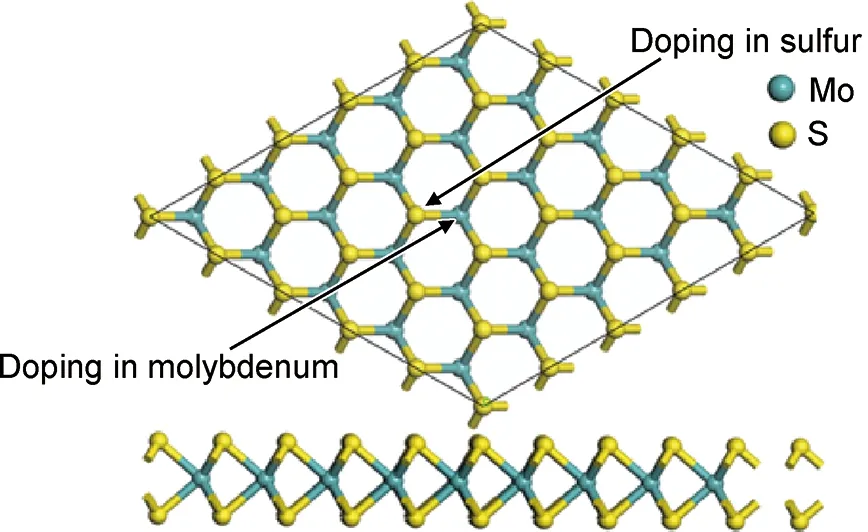
图1 掺杂MoS2俯视图和侧视图Fig.1 Top view and side view of the doped MoS2
2 结果与讨论
2.1 Cu, Ag, Au掺杂MoS2体系的杂质能及键长畸变
对于掺杂体系,掺杂位置的不同其稳定性也是不一样的,所以分别计算了Mo位和S位的掺杂总能和杂质替换能,最后以能量极小值为判据得到吸附和掺杂的稳态位置。杂质替换能ΔE为[14]:
(1)
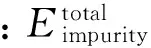
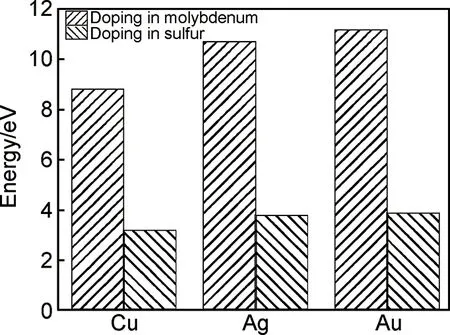
图2 Cu,Ag,Au掺杂杂质替换能Fig.2 Impurity replacement energy of Cu,Ag and Au doped MoS2
表1给出了超胞MoS2和贵金属掺杂在S位的超胞几何结构优化后的计算结果。在S位掺杂时,dA-S代表A(A= S, Cu, Ag, Au) 原子与其最近邻S原子的键长,dA-Mo代表A 原子与其最近邻Mo原子的距离。对于Cu, Ag, Au在S位掺杂,杂质与最近邻的Mo,S原子的键长都发生了畸变,Cu与最近邻的Mo,S原子的键长分别为0.2660,0.3065nm;Ag与最近邻的Mo,S原子的键长分别为0.2934,0.3117nm;Au与最近邻的Mo,S原子的键长分别为0.3006,0.3180nm;相对于未掺杂结构,畸变率最大的是dAu-Mo,达23.8%。
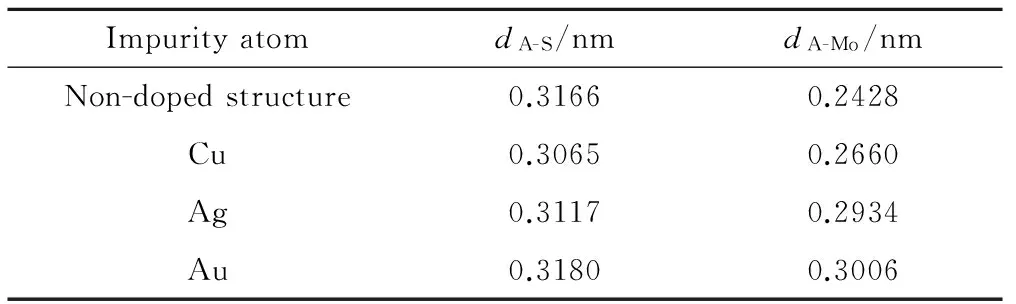
表1 未掺杂和掺杂MoS2体系的键长
2.2 Cu, Ag, Au在S位掺杂MoS2体系的能带结构及态密度
为了分析Cu, Ag, Au在S位掺杂MoS2体系的能带结构的变化情况,本工作将单层 MoS2超胞结构和掺杂的能带结构进行了对比,如图3所示。能量范围为-2.0~2.0eV。可以看出, 单层 MoS2的和Cu, Ag, Au掺杂体系的价带顶与导带底都在布里渊区Q点,表明掺杂也为直接带隙材料; 掺杂体系的禁带宽度都在1.7eV左右,掺杂对其禁带的宽度影响不大。与单层 MoS2的超胞相比,掺杂体系的禁带中出现了新能级, Cu掺杂体系禁带区中有4个能级,一条位于费米能级,其余3条分别位于0.13,0.22,0.26eV处,Ag掺杂的禁带区域中有4个能级, 分别位于0,0.125,0.21,0.28eV处, Au掺杂的禁带中也只有4个能级,分别位于0,0.125,0.20,0.25eV;同时掺杂体系的导带和价带都不同程度地向低能区域移动,Cu, Ag, Au在S位掺杂的导带能量都低于1.25eV。其中下降最多的是Au掺杂的体系,导带底和价带顶的能量值分别降低了约0.7,0.95eV;然而杂质Cu, Ag, Au的3个原子的电子组态中都存在d态电子,掺杂体系的能量值的下降与d态电子有关。

图3 能带结构 (a)单层MoS2;(b)Cu掺杂;(c)Ag掺杂;(d)Au掺杂Fig.3 Band structures (a)monolayer MoS2;(b)Cu doped MoS2;(c)Ag doped MoS2;(d)Au doped MoS2
图4给出了单层 MoS2的超胞以及Cu,Ag,Au在S位掺杂体系的总态密度和分态密度。为了便于对比分析,统一选取的能量范围为-15~2.5eV之间, 其中包含了上、下价带和导带的全部分布信息。图4(a),(b),(c),(d)分别对应于超胞结构以及Cu,Ag,Au 掺杂MoS2的计算结果。可以看出,MoS2的超胞以及掺杂MoS2体系的导带主要分布在0.75~1.25eV之间;MoS2的超胞结构导带主要分布在1.79~2.5eV之间。上、下价带的能量主要由Mo4d和S3p电子贡献;而导带部分的能量状态仍以Mo4d和S3p电子贡献为主, 同时也存在杂质原子最外层5d和5S电子贡献。
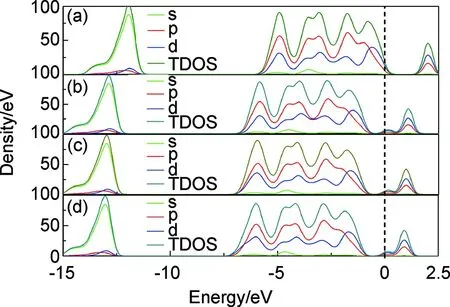
图4 态密度 (a)单层MoS2;(b)Cu掺杂;(c)Ag掺杂;(d)Au掺杂Fig.4 Density of states (a)monolayer MoS2;(b)Cu doped MoS2;(c)Ag doped MoS2;(d)Au doped MoS2
2.3 Cu, Ag, Au在S位掺杂MoS2体系的电荷分布
为了研究Cu, Ag, Au掺杂对MoS2电荷的分布影响,分析了单层超胞MoS2中S,Mo原子的电荷,可知二者分别为-0.199,0.393。对于Au掺杂MoS2体系,Au原子的电荷为0.456, 近邻的Mo原子的电荷为0.252,近邻的S原子的电荷为-0.198;对于Ag掺杂MoS2体系,Ag原子的电荷为0.438, 近邻的Mo原子的电荷为0.275,近邻的S原子的电荷为-0.203;对于Cu掺杂MoS2体系,Cu原子的电荷为0.329, 近邻的Mo原子的电荷为0.300,近邻的S原子的电荷为-0.211。所以,Cu,Ag,Au掺杂MoS2体系中,杂质原子周围存在着电荷聚集,同时也存在电荷损失。
3 结论
(1)Cu,Ag,Au在S位掺杂的杂质能都低于在Mo位掺杂的杂质能,表明在S位掺杂的体系的稳定性强于在Mo位掺杂的体系。
(2)Cu,Ag,Au在S位掺杂,杂质与最近邻的Mo,S原子的键长都发生了畸变,畸变率最大的是dAu-Mo,达23.8%。
(3)与单层 MoS2的超胞相比,掺杂体系的禁带中出现了4条新能级,掺杂体系的能量向低能区移动与d态电子有关。
(4)Cu, Ag, Au掺杂MoS2体系中,杂质原子周围存在着电荷聚集,同时也存在电荷损失。
[1] 黄宗玉.类石墨烯二硫钼的第一性原理研究[D].湘潭:湘潭大学,2014.
HUANG Z Y.First-principles study of graphene-like molybdenum disulfide[D].Xiangtan:Xiangtan University,2014.
[2] 范梦慧,蔡勋明,岑伟富,等.单层MoS1.89Xo.11电子结构及光学性质的第一性原理计算[J].激光与光电子学进展,2015,52(5):163-170.
FAN M H,CAI X M ,CEN W F,et al.First-principles calculation of electronic structure and optical properties of monolayer MoS1.89-Xo.11[J].Laser & Optoelectronics Progress,2015,52(5):163-170.
[3] FENG L P,SU J,LIU Z T.Effect of vacancies on structural,electronic and optical properties of monolayer MoS2:a first-principles study[J].Journal of Alloys Compounds,2014,613(10):122-127.
[4] WANG Y Z,WANG B L,HUANG R,et al.First-principles study of transition-metal atoms adsorption on MoS2monolayer[J].Physica E:Low-dimensional Systems and Nanostructures,2014,63(9):276-282.
[5] LATE D J,LIU B,MATTEH S,et al.Hysteresis in single-layer MoS2field effect transistors[J].ACS Nano,2012,6(6):5635-5641.
[6] SUNGH N,JABBOUR G,SCHWINGENSCHOGL U.Optical and photocatalytic properties of two-dimensional MoS2[J].Physics of Condensed Matter,2012,85(11):1-4.
[7] 张昌华,余志强,廖红华.Te掺杂单层MoS2电子结构与光电性质[J].发光学报,2014,35(7):785-790.
ZHANG C H,YU Z Q,LIAO H H.Electronic structure and photoelectric properties of Te-doped single-layer MoS2[J].Chinese Journal of Luminescence,2014,35(7):785-790.
[8] 吴木生,徐波,刘刚,等.Cr和W掺杂的单层MoS2电子结构的第一性原理研究[J].物理学报,2013,62(3):037103-6.
WU M S,XU B,LIU G,et al.First-principles study on the electronic structures of Cr-and W-doped single-layer MoS2[J].Acta Phys Sin,2013,62(3):037103-6.
[9] 曹娟,崔磊,潘靖.V,Cr,Mn掺杂MoS2磁性的第一性原理研究[J].物理学报,2013,62(18):187102-7.
CAO J,CUI L,PAN J.Magnetism of V,Cr and Mn doped MoS2first-principal study[J].Acta Phys Sin,2013,62(18):187102-7.
[10] 雷天民,吴胜宝,张玉明,等.La,Ce,Nd掺杂对单层MoS2电子结构的影响[J].物理学报,2014,63(6):067301-8.
LEI T M,WU S B,ZHANG Y M,et al.Effects of La,Ce and Nd doping on the electronic structure of monolayer MoS2[J ].Acta Phys Sin,2014,63(6):067301-8.
[11] 刘俊,梁培,舒海波,等.单层MoS2分子掺杂的第一性原理研究[J].物理学报,2014,63(11):117101-7.
LIU J,LIANG P,SHU H B,et al.First principles study on molecule doping in MoS2monolayer[J].Acta Phys Sin,2014,63(11):117101-7.
[12] MAO R,KONG B D,KIM K W.Thermal transport properties of metal/MoS2interfaces from first principles[J].Journal of Applied Physic,2014,116(3):42-47.
[13] PERDEW J P,BURKE K,ERNZERH M.Generalized gradient approximation made simple[J].Phys Rev Lett,1996,77(18):3865-3868.
[14] 张培新,陈建华,魏群.掺杂材料分子模拟与计算[M].北京:科学出版社.2012.131.
ZHANG P X,CHEN J H,WEI Q.Calculation of Molecular and Doped Materials[M].Beijing:Science Press,2012.131.
[15] 甄文柱,梁波.等离子喷涂MoS2/Cu基复合涂层真空摩擦磨损性能[J].材料工程,2013,(8):16-22.
ZHEN W Z,LIANG B.Tribological behavior of plasma sprayed MoS2/Cu composite coating under vacuum atmosphere[J].Journal of Materials Engineering,2013,(8):16-22.
First Principles Calculation of Electronic Structure of Doped Monolayer MoS2
FU Chun-ping1,2
(1 Engineering Research Center of Electronic InformationTechnology and Application,Chongqing University of Arts and Sciences,Chongqing 402160,China;2 Chongqing Key Laboratory of Micro/Nano Materials Engineering and Technology,Chongqing 402160,China)
Based on the first principle, the bond length, band structures and density of states of Cu, Ag and Au doped monolayer MoS2were studied. The effects of Cu, Ag and Au doped on the monolayer MoS2electronic structure were discussed. The results show that the impurity energy of Cu, Ag, Au doping at the S position always lower than that doping at the Mo position, so that there is the more stably of Cu, Ag, Au doping at the S position. When the Cu, Ag, Au dope at the S position, the bond length of impurity atom and nearest neighbor S, Mo atom are distorted, the maximum of aberration ratedAu-Mois up to 23.8%.Comparing with the monolayer MoS2,the doped system appears four new energy levels in forbidden band. The energy of the conduction band and valence band turns to the low energy region.Charge gathers around impurity atoms,at the same time,a charge loss exits.
MoS2;band structure;density of states;doped;first principle
10.11868/j.issn.1001-4381.2016.12.013
O469
A
1001-4381(2016)12-0080-04
重庆文理学院校级科研项目资助(Z2015DQ07);永川区科委项目资助(Ycstc,2014nc4002);重庆市科学技术委员会基础与前沿研究计划资助项目(cstc2014jcyjA00042);重庆市教委科技项目资助(KJ1601128);重庆市高校微纳米材料工程与技术重点实验室(KF2016012)
2015-08-21;
2016-07-11
伏春平(1986-),男,硕士,主要从事二维材料的物性研究工作,联系地址:重庆市永川区红河大道319号重庆文理学院电子信息技术与应用工程中心(402160),E-mail:fuchunping@163.com
