C3启动子克隆及荧光素酶报告基因载体构建
2016-12-15SalmanNaseerShahidKhanImran何冰林佳
Salman Naseer,Shahid Khan,M Imran,何冰,林佳
C3启动子克隆及荧光素酶报告基因载体构建
Salman Naseer1,Shahid Khan1,M Imran1,何冰2,林佳2
(华北理工大学 1. 国际教育中心,2. 生命科学学院,河北 唐山 063000)
为克隆C3基因启动子序列,构建C3基因启动子荧光素酶报告基因载体,运用PCR的方法从血液中提取到的基因组DNA中获得目的基因,双酶切后连接到荧光素酶报告基因载体上,构建C3基因启动子报告基因载体.构建的pGL3-Basic-C3-promotor重组质粒和内参质粒SV40瞬时转染Hela细胞,检测荧光素酶活性.结果表明,PCR扩增出C3基因启动子片段;双酶切及测序鉴定重组体构建正确;转染Hela细胞后经双荧光素酶报告基因检测确定重组体有启动子活性.成功构建了C3启动子报告基因载体,在细胞系中进行初步活性分析得知所克隆的C3基因调控序列包含启动子活性区域.
C3;启动子;荧光素酶报告基因;载体构建
人补体第三组分(Complement 3,C3)是补体系统的核心蛋白质,它是1912年Ritg用蛇毒处理血清时发现的[1].在补体各成分中,C3在血清中含量最高;在功能上,C3亦居于中心地位,它既是几条激活途径的交汇,又是C3b依赖性阳性反馈环路的基础;同时,C3裂解片段及其结合蛋白复杂而多样,在免疫防御、免疫调控以及免疫病理中发挥着重要作用[2].研究发现,C3的表达受到营养状况、激素水平和发展阶段等生理因素的调控,且在多种肿瘤中也发现了C3表达水平的差异[3].本实验旨在寻找人C3基因上游调控序列启动子区域,为深入研究人C3基因转录调控机制提供实验基础.
1 材料与方法
1.1基因组DNA的提取
由实验参与人员提供人血液样,采用人全血基因组DNA提取试剂盒(天根公司),按照说明书方法进行DNA提取.结果用1%琼脂糖凝胶电泳检测DNA纯度及完整性,利用分光光度计测定产物在260 nm处的吸收值,以计算浓度.
1.2C3启动子的PCR扩增
根据GenBank报道的C3启动子序列,已知人的C3基因启动子序列设计两端引物上下游引物分别加入Nhe I和Hind Ⅲ 2个酶切位点.上游引物为:5′-ATACAGAGGAGTGAGCAGGTAAAG-3′(下划线部分为Nhe I酶切位点),下游引物为:5′-GATAGTTCCCACCGCATTT-3’(下划线部分为Hind Ⅲ酶切位点),PCR引物由北京诺赛基因组研究中心合成,PCR反应条件为预变性95 ℃ 2 min,94 ℃ 25 s,59 ℃ 1 min 40 s,72 ℃ 25 s,72 ℃ 5 min,35个循环,4 ℃保温.PCR产物大小为2 513 bp.
1.3重组质粒的构建
用pGL3-Basic载体(见图1)质粒转化常规制备的大肠杆菌DH5感受态细胞,进行大量扩增,用质粒提取试剂盒(Promega公司)大量制备pGL3-Basic质粒,用ScanDrop 200(analytik Jena,德国)测定pGL3-Basic质粒的纯度和浓度.分别用限制性内切酶Nhe I和Hind Ⅲ对胶回收的C3启动子PCR产物以及pGL3-Basic质粒载体进行双酶切4 h,胶回收酶切产物.将胶回收纯化过的C3启动子片段和pGL3-Basic质粒载体在16 ℃连接过夜.
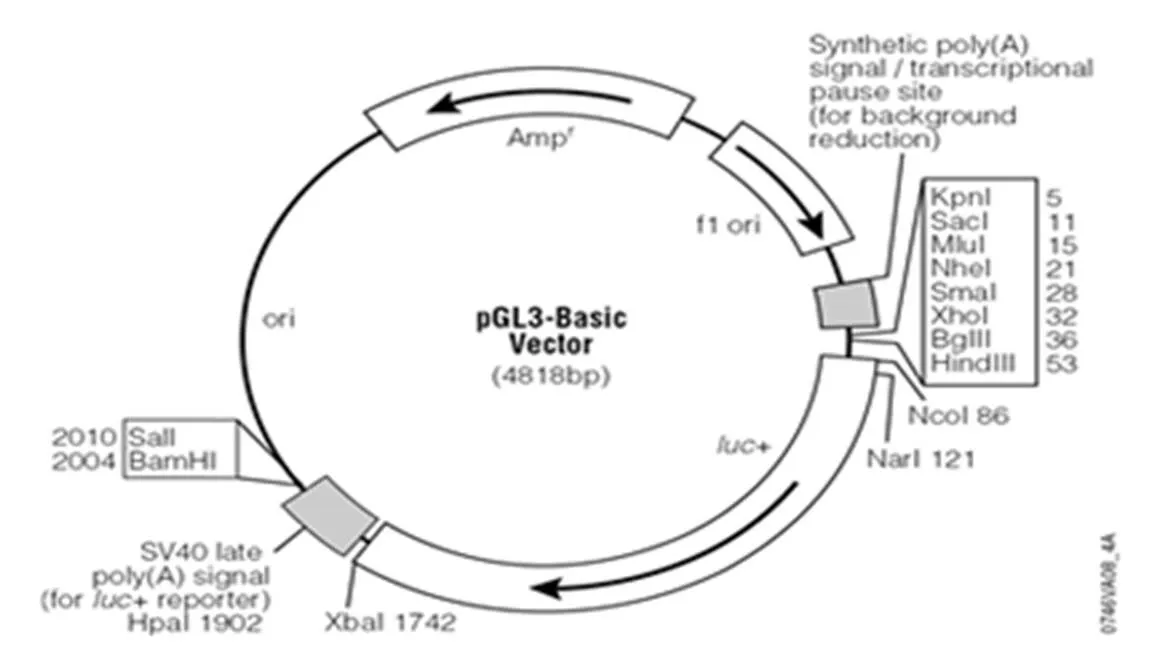
图1 pGL3-Basic载体图谱
取10 μL连接产物转化100 μL大肠杆菌DH5感受态细胞.经过含有氨苄的LB固体培养基中过夜培养,从转化子的平板上随机挑选10个单菌落,经含有氨苄抗性的液体LB培养基扩增培养后用质粒抽提试剂盒提取质粒,进行酶切和测序鉴定.
1.4C3启动子活性的鉴定
将生长状态良好的Hela细胞按1×104个细胞/孔的量在96孔板中接种,当细胞融合度达到60%之后,采用脂质体DNA转染法将重组质粒、内参载体质粒pRL-SV40共转染Hela细胞,设立空白载体为对照,经过一定时间的培养后收集,吸弃培养基,用PBS洗1次,加入裂解液后室温摇动10 min.按照双荧光素酶检测试剂盒(Dual Luciferase Assay System)操作说明书,利用化学发光仪测定萤火虫荧光素酶发光值,并以空载体实验组的比值做对照,分析启动子活性.每组设立3个复孔,每次实验重复3次,得到3个相对值,取其平均值.
2 结果
2.1C3启动子序列的PCR扩增
以人基因组DNA为模板,用制备的引物进行PCR扩增.产物在1%的琼脂糖凝胶中进行电泳,结果见图2.扩增出的条带位于2 513 bp位置,无非特异扩增,经测序鉴定为目的条带.
2.2C3启动子荧光素酶报告基因载体的双酶切鉴定
酶切后重组体琼脂糖凝胶电泳图谱见图3.由图3可见,经限制性内切酶Nhe I和Hind Ⅲ双酶切后琼脂糖凝胶电泳显示,重组体在4.8 kb(载体片段位置)及2 500 bp左右(目的片段位置)各出现1条相应的条带,进一步证明了C3启动子序列已经插入pGL3-Basic质粒中.

图2 C3启动子PCR产物电泳图
注:M为marker;1,2泳道为PCR产物.

图3 酶切后重组体琼脂糖凝胶电泳图谱
注:M为marker;1泳道为酶切产物.
2.3C3启动子荧光素酶报告基因载体的测序结果
用pGL3-Basic多克隆区域通用引物,北京诺赛基因组中心测序结果经NCBI的Blast显示,pGL3-Basic质粒中插入的片段大小为2 513 bp,并且序列与C3启动子序列完全一致(见图4),表明pGL3-Basic-C3载体构建成功.

图4 C3启动子片段测序结果
2.4C3启动子荧光素酶报告基因载体活性分析
将重组体pGL3-Basic-C3-promotor与内参质粒pRL-SV40共转染Hela细胞,48 h后收集细胞检测荧光素酶活性,并将两者比值作为衡量C3启动子活性的依据,SV40作为内参可以消除由于细胞数目及转染效率等不同所带来的组间差异.3次独立重复实验的统计图见图5.由图5可见,pGL3-Basic-C3所检测出的荧光素酶活性是对照组pGL3-Basic的5.3倍(=0.01),此结果提示C3片段有启动子活性.

图5 pGL-Basic-C3-promoter启动子活性检测
注:<0.05.
3 讨论
报告基因是研究基因功能、基因与基因间调节、蛋白与基因作用以及非编码对基因表达调控的重要手段.目前,双荧光素酶报告系统是应用最为广泛的报告基因系统.荧光素酶由于其表达量与发光强度相关,因而可以作为一种报告酶并被应用于测定原核及真核细胞中的基因表达量及启动子活性.应用比较广泛的双荧光素酶报告载体——pGL3-Basic载体,可以准确快速地连接克隆靶基因,并使用特定检测荧光素酶试剂盒和检测仪来进行检测[4].因此,可使用该报告基因质粒载体对人肿瘤组织或肿瘤细胞中重要的补体基因C3启动子活性进行检测,并作为监测肿瘤发生发展的重要手段.
本研究采用基因重组技术将C3基因启动子连接到具有多克隆位点的pGL3-Basic载体质粒上,构建能够瞬时表达C3活性的重组质粒,为研究抗肿瘤药物的作用靶点提供了实验平台.同时检测萤火虫荧光素酶的活性来反应C3基因启动子的转录活性,可应用于批量筛选以C3基因为靶向的抗肿瘤药物,具有靶点明确、高效快速及反应灵敏等特点.但本研究并没有进行C3启动子的功能分析,需要在后续实验中继续进行此方面的研究.
[1] Manning G,Whyte D B,Martinez R,et al.The protein kinase complement of the human genome[J].Science,2002,298(5600):1912-1934
[2] Cravedi P,Heeger P S.Complement as a multifaceted modulator of kidney transplant injury[J].Journal of Clinical Investigation,2015,125(3):1365-1365
[3] Cheung N K,Walter E I,Smith-Mensah WH,et al.Decay-accelerating factor protects human tumor cells from complement-mediated cytotoxicity in vitro[J].Journal of Clinical Investigation,1988,81(4):1122-1128
[4] Liu X J,Kong X X,Wang R,et al.Construction of pGL3-Basic-SREBP-1c-promoter reporter gene vector and detection of its function[J].Basic & Clinical Medicine,2009,29(5):495-498
Cloning of human C3promoter and construction of the promoter luciferase reporter gene vectors
Salman Naseer1,Shahid Kham1,M Lmran1,HE Bing2,LIN Jia2
(1. International Education College,2. College of Life Science,North China University of Science and Technology,Tangshan 063000,China)
To clone the human C3gene promoter,and to construct the luciferase reporter gene vector containing the C3gene promoter sequence. The C3promoter fragment was amplified from genomic DNA by PCR. After digestion with restriction enzymes,the C3promoter was cloned into the luciferase reporter vectors. Then the recombinant vector was transiently transfected into Hela cell with pRL-SV40. The activity of luciferase was detected 48 hours later. The results showed that C3gene promoter,a fragment about 2 513bp,was amplified by PCR. Double digestion and sequencing revealed that the recombinant vector had a promoter activity. Conclusion the human C3promoter luciferase reporter gene vector was successfully constructed. The cloned C3gene fragment was demonstrated to contain the cis-regulatory elements.
C3;promoter;luciferase reporter gene vector;vector construction
1007-9831(2016)11-0041-04
Q78
A
10.3969/j.issn.1007-9831.2016.11.011
2016-09-15
华北理工大学大学生创新创业训练计划项目(X2015257)
Salman Naseer(1990-),男,巴基斯坦旁遮普人,在读本科留学生.E-mai:Dr_Salmankhan@ymail.com
林佳(1982-),女,河北唐山人,讲师,硕士,从事分子生物学研究.E-mail:linjia825@126.com
