Al掺杂纤锌矿GaN电子结构第一性原理计算
2016-12-15陈俊利尹海涛郑青松
陈俊利,尹海涛,郑青松
(哈尔滨师范大学)
Al掺杂纤锌矿GaN电子结构第一性原理计算
陈俊利,尹海涛*,郑青松
(哈尔滨师范大学)
采用第一性原理方法,在LMTO-MBJ框架下精确的计算GaN和AlN的电子结构,计算结果与实验值一致.应用CPA的方法计算任意量的Al掺杂GaN合金的电子结构,理论计算表明,GaN的带隙宽度随Al掺杂浓度x的增大而增大,而且满足关系式Eg=3.43+2.3x(0≤x≤0.65),Eg=2.45+3.7x(0.65≤x≤1).计算可为氮化物半导体GaN、AlN及其三元合金化合物Ga1-xAlxN的实验研究提供理论依据.
GaN;AlN;第一性原理;带隙
0 引言
近些年来,IIIA-VA族氮化物半导体GaN、AlN及其三元合金化合物Ga1-xAlxN在微电子学、光电子学和现代通讯等领域拥有广阔的应用前景,使之成为国内外科研人员研究的热点.相比于以Si、Ge为代表的第一代半导体和以GaAs、InP为代表的第二代半导体,GaN、AlN及Ga1-xAlxN具有宽的直接带隙、有较大的击穿场能、高的热导率、高的光电转换效率、较大的饱和电子速度和稳定的化学性质等优异性能,已成为第三代半导体材料的典型代表.
Ga1-xAlxN作为合金半导体,通过改变Al和Ga的组分,其禁带宽度可以从3.45eV( GaN )到6.20eV ( AlN)之间连续可调[1],覆盖了从可见光区到紫外光区的范围.优异的物理化学性能,使其在绿、蓝、紫、紫外及白光发光二极管(LED);激光二极管(LD);紫外探测器等领域有着广泛的应用前景[2].同时,纳米Ga1-xAlxN有尺寸相关的光电性能,在介观物理研究和制造纳米器件方面也具有潜在的应用价值.同其它半导体材料一样,对Ga1-xAlxN的掺杂也成为研究其应用的基础,成为近两年研究的前沿与热点.近年来科研人员陆续利用分子束外延法(MBE)[3]、金属有机物化学气相沉积法(MOCVD)[4]、磁控溅射法等技术成功制备出了Ga1-xAlxN材料;理论方面,国内外也进行了一些对GaN和A1N材料结构和性能的模拟计算,然而,对三元半导体Ga1-xAlxN及其掺杂的理论研究却鲜有报道.
该文工作精确的计算了GaN和AlN的带隙宽度.在GaN中掺Al可形成Ga1-xAlxN合金,通过控制Al的掺杂量能任意有效地调节Ga1-xAlxN合金的禁带宽度,由初始禁带宽度3.45eV增加到6.20 eV.为研究IIIA-VA族氮化物GaN、AlN及其三元合金化合物Ga1-xAlxN提供理论依据.
1 计算方法
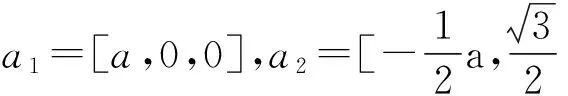
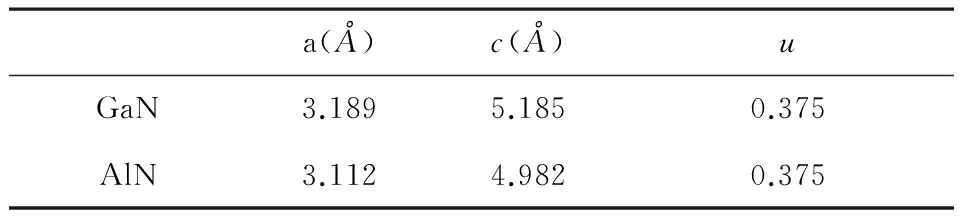
表1 纤锌矿GaN和AlN 晶格常数
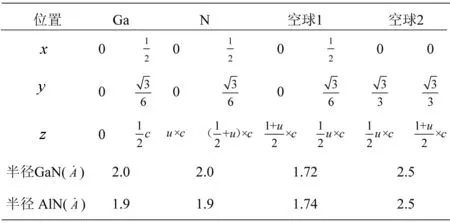
表2 GaN、AlN原子球和空球的位置和半径
2 计算结果和分析
正确的电子结构是理论预测的前提,相比较广义梯度近似(GGA)和局域态密度近似(LDA),应用MBJ半局部交换势可准确计算半导体材料的能带结构[8],其具体公式为:
(1)

我们首先计算纤锌矿GaN、AlN的电子结构.计算的能带结构如图1所示,MBJ方法计算的带隙宽度是3.45eV和6.20eV,与LDA和GGA计算的带隙宽度相比较,带隙变宽非常明显,并且和实验值一致.
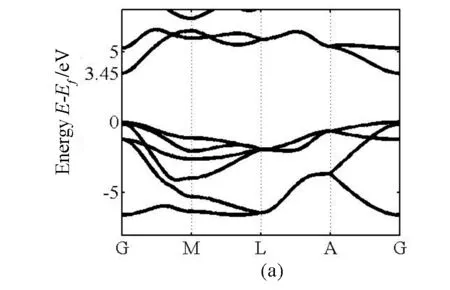
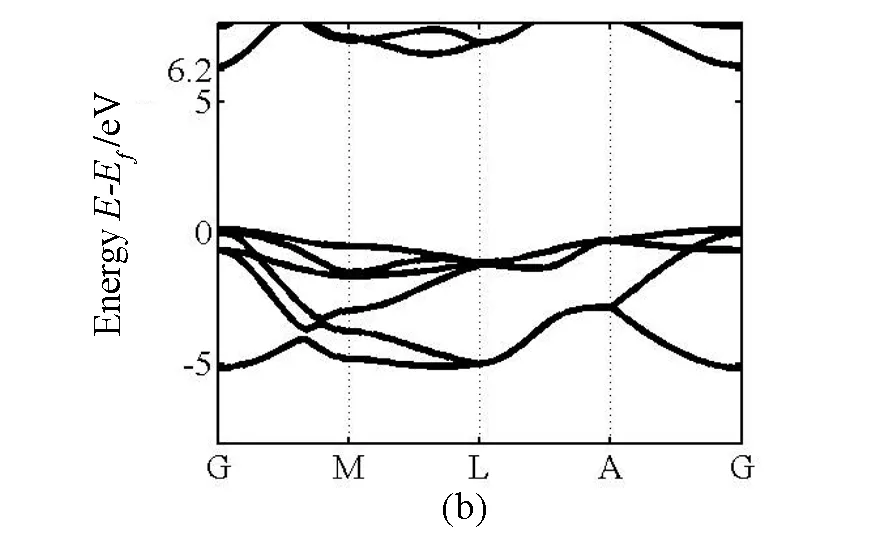
图 1 (a)GaN能带结构,计算带隙值为3.45 eV; (b)AlN能带结构,计算带隙值为6.20 eV
确定纯GaN、AlN带隙宽度后,可以利用CPA-MBJ方法计算任意浓度Al掺杂GaN合金的电子结构,合金Ga1-xAlxN 的带隙宽度能够通过态密度(DOS)估算出,DOS是动量k和能量 的函数,可由推迟格林函数虚部给出:
(2)

(3)
利用公式(2)计算的合金Ga0.5Al0.5N的态密度如图2所示.计算的带隙宽度为4.56eV,比纯GaN带隙宽度(3.45eV)高1.11eV.BergmannMJ等人测量了Ga0.5Al0.5N合金的带隙值[9],他们测量的带隙宽度为4.50eV.和我们的计算结果基本一致.

图2 Ga0.5Al0.5N的态密度图,计算带隙值为4.56 eV
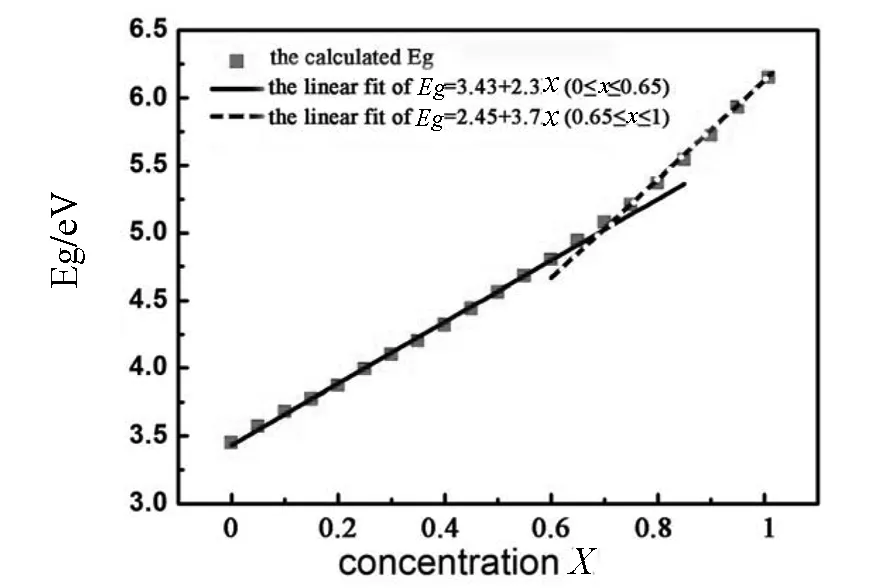
图3 纤锌矿Ga1-xAlxN的带隙宽度随Al掺杂浓度x的变化,红色方块是理论计算值,实线和虚线是线性拟合的效果
在以往的研究工作中,大多低估GaN、AlN及其合金的带隙值,而且,在计算Ga1-xAlxN合金性质时,通过构建超级晶胞并用Al替代Ga原子的方法来研究不同掺杂浓度的GaN性质,这种方法只能研究一些特殊浓度的掺杂,并且浓度不能取很低,否则需要构建很大的晶胞,需要很大的计算量.利用CPA方法可避免这一问题,可以利用原胞计算任意浓度的掺杂问题,既减少了计算量,也便于总结Ga1-xAlxN带隙宽度的变化与任意浓度的Al掺杂之间的关系.图3给出了带隙宽度随Al掺杂浓度变化的情况,浓度变化范围取0≤x≤1,浓度变化间隔为0.05.如图3所示,带隙宽度随Al的掺杂浓度线性的增加.当0≤x≤0.65时满足关系式Eg=3.43+2.3x,当0.65≤x≤1时满足关系Eg=2.45+3.7x.从电子结构的观点来看,导带底的位置主要是由Ga的4s态电子决定的,而价带顶主要由N的2p态电子决定的.当Al掺杂到GaN晶体中,价带顶的位置还是由N的2p态电子决定,且位置基本保持不变,导带底的位置也仍然由Ga的4s态电子决定,但它向高能方向微偏移,从而引起光学带隙宽度变大[10].
3 结束语
该论文采用DFT-NEGF方法,在LMTO-MBJ-CPA框架下,在精确确定纤锌矿结构GaN、AlN的电子结构后,研究不同浓度的Al掺杂GaN合金的电子结构,得到了GaN带隙宽度随Al掺杂浓度变化关系.研究表明随着Al的掺杂浓度的增加导致GaN晶体的禁带宽度增大,并且满足关系式Eg=3.43+2.3x(0≤x≤0.65),Eg=2.45+3.7x(0.65≤x≤1).该文计算结果可为实验研究III-V族氮化物GaN、AlN及其三元合金化合物Ga1-xAlxN提供理论基础.
[1] Wang L, Hao Z B, Han Y J, et al. Response time improvement of AlGaN photoconductive detectors by adjusting crystal-nuclei coalescence process in metal organic vapor phase epitaxy[J] .Journal of Semiconductors, 2011,32(1):014013.
[2] Pecora E F, Zhang W, Nikiforov A Y ,et al. Sub-250nm light emission and optical gain in,AlGaN materials[J]. Journal of Appl Phy,2013, 113(1):013106.
[3] Novikov S V, Staddon C R., Powell R. E L,et al. Wurtzite Ga1-xAlxN bulk crystals grown by molecular beam epitaxy[J],. Journal of Crystal Growth, 2011,322(1):23-26.
[4] Pan X, Wang X L, Xiao H L ,et al. Raman study on dislocation in high Al content AlxGa1-xN [J] .Eur Phy J-Appl Phys, 2012, 58(1):10102.
[5] Maassen J, Harb M, Michaud-Rioux V, Zhu Y, and Guo H, et al.Quantam transport modeling from first principle.Proc IEEE,2013:101-518.
[6] Zhu Y, Liu L, and Guo H, et al .Quantum transport theory with the nonequilibrium coherent potentials. Phys Rev B,2013,88:205415.
[7] Kudrnovsky J, Drchal V, and Masek J, et al . Coherent-potential approximation in the tight-binding linear muffin-tin orbital method[J]. Phys Rev B 1987,35:2487.
[8] Tran F and Blaha P, et al .Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential[J].Phys Rev Lett. 2009,102:226401.
[9] Bergmann M Jand Casey H C, et al .Optical-field calculations for lossy multiple-layer AlxGa1-xN/ InxGa1-xN laser diodes[J]. Appl Phys,1998,84:1196.
[10] Guo J Y,Zhen G ,He K H and Chen,et al . First-principle study on electronic structure and optical properties of Al and Mg doped GaN J Z[J]. Acta Phys Sin, 2008,57:3740-07.The Research on First-principles Electronic Structure for Al-doped Wurtzite GaN
(责任编辑:季春阳)
Chen Junli, Yin Haitao, Zheng Qingsong
(Harbin Normal Univerisyt)
Using first-principles approach, the band gap of the wurtzite GaN,AlN and band gap of the ternary alloy Ga1-xAlxN are studied, where the modified Becke-Johnson (MBJ) semi-local exchange is used to accurately determine the band gap, and the coherent potential approximation (CPA) is applied to deal with confi gurational average for the ternary alloys. The band gap of Ga1-xAlxN increasing with the concentration of Al are found. The calculated band gaps the ternary alloy Ga1-xAlxN satisfied the relationship betweenEg=3.43+2.3x(0≤x≤0.65),Eg=2.45+3.7x(0.65≤x≤1). These theoretical prediction will be useful in the design and application of Ga1-xAlxN device.
GaN;AlN;First principle;Band gaps
2016-09-22
O47
A
1000-5617(2016)03-0063-03
*通讯作者
