基于高通量测序技术分析北京清香型大曲微生物多样性
2016-12-14张双燕廖永红纪南周晓宏
张双燕,廖永红,纪南,周晓宏*
(1.北京理工大学生命学院,北京100081;2.北京工商大学食品学院,北京100048)
基于高通量测序技术分析北京清香型大曲微生物多样性
张双燕1,廖永红2,纪南2,周晓宏1*
(1.北京理工大学生命学院,北京100081;2.北京工商大学食品学院,北京100048)
利用高通量测序方法对北京某清香型酒厂大曲细菌16S rDNA V4区和真菌ITS1区进行测序分析其微生物多样性,并对大曲微生物在不同分类水平相对丰度进行统计。结果表明:大曲中在细菌操作分类单元(OTU)为47、真菌OTU为11,细菌种类较真菌种类丰富;细菌中优势菌是乳酸菌,包括乳杆菌属、片球菌属、乳球菌属、明串珠菌属四个属。同时也检测到醋杆菌属、芽孢杆菌属等;真菌中优势菌是假丝酵母属,此外还有曲霉属、毛霉属、威克汉姆酵母属等;对大曲物种Alpha多样性分析表明测序深度基本覆盖该酒曲中微生物种类;最后分析讨论了大曲优势微生物在白酒酿造中的功能。该研究对于指导白酒生产具有重要意义。
清香型大曲;高通量测序;微生物;16S rDNA V4;内转录间隔区
我国白酒历史悠久,是世界七大蒸馏酒之一。中国白酒传统生产方式是固态发酵,用曲酿酒是我国白酒生产的特色,在世界酿酒史上独树一帜。日本学者板口谨一郎先生认为,中国创造酒曲酿酒,是我国劳动人民智慧的结晶,其重要性可与中国四大发明媲美[1-2]。酒曲是以粮食为原料制成的物系、酶系、菌系共存的糖化发酵剂。白酒生产实质上是多种微生物共同作用的结果,酒曲作为白酒生产中微生物的主要来源,其中的微生物按照功能可分为糖化微生物、酿酒微生物和产香微生物三大类[3]。
最初研究白酒微生物的方法多为传统微生物分离培养法。利用传统方法分离到的微生物在种类和数量上是有限的,相比之下,分子生物技术则成为微生物研究强有力的工具。近年来,聚合酶链式反应和变性梯度凝胶电泳结合(polymerase chain reaction-denatured gradient gel electrophoresi,PCR-DGGE)技术、DNA克隆文库、高通量测序等方法逐渐被应用于白酒微生物研究。WANG H Y等[4]利用PCR-DGGE方法分析了10种酒曲的微生物类群,并对其进行比较,证明大曲微生物群落组成具有地区差异性。陈玲等[5]运用16S rDNA克隆文库法与高通量测序法对比分析了浓香型大曲微生物群落结构,两种方法各具优势,在分析优势菌群上结果基本相同,但高通量测序法分析到的微生物类群更多。
高通量测序技术[6-7]作为新一代测序方法,在微生态领域有广泛的应用前景,该技术具有通量高、成本低、准确率高、可进行双向测序等优点,可以方便快捷地分析样品中复杂的微生物菌群结构,是分析复杂多样微生物样品的首选。酒曲微生物非常复杂、种类繁多,更需要从微生态角度探究微生物的多样性。2015年,乔晓梅[8]利用高通量测序技术分析了清香大曲中真菌群落结构,但只在种水平上分析了真菌组成,并没有在其他分类水平上分析真菌;雷振河[9]基于高通量测序技术分析了山西某酒厂大曲和酒醅中微生物群落结构,对于大曲,只有细菌门分类水平和真菌属分类水平的分析;两者对大曲微生物的分析都只局限于某一个分类水平,而没有全面地从门、纲、目、科、属、种等各个分类水平上分析微生物多样性。本研究以北京某酒厂大曲微生物为研究对象,对其进行16S rDNA和内转录间隔区(internal transcribed spacer,ITS)可变区域高通量测序,同时分析酒曲细菌和真菌的多样性,并全面系统地统计分析酒曲微生物在门、纲、目、科、属、种不同分类水平上的相对丰度。酒曲微生物多样性分析对于指导筛选白酒酿造过程中功能微生物有重要意义。
1 材料与方法
1.1 材料与试剂
1.1.1 材料
大曲样品:取自北京某清香型白酒酒厂。
1.1.2 主要试剂
氯仿、异戊醇、无水乙醇、异丙醇(分析纯):北京化工厂;Tris-酚:美国Sigma公司;蛋白酶K:德国Merck公司;RNA酶:日本TaKaRa Biotechnology公司。
1.2 仪器与设备
ProS聚合酶链式反应(polymerasechainreaction,PCR)仪:德国Eppendorf公司;PowerPac3000电泳仪:美国Bio-Rad公司;Miseq高通量测序平台:美国Illumina公司。
1.3 实验方法
1.3.1 酒曲样品准备
自酒厂曲房内大曲的表层、中间、底部分别取样约10 g并粉碎至合适大小后混匀放于无菌袋中,于-20℃保存备用。
1.3.2 基因组DNA的提取
采用盐析改进法[10]提取大曲中微生物基因组DNA,具体可分为大曲预处理、核裂解液和蛋白酶K裂解、酚/氯仿/异戊醇的抽提、异丙醇的沉淀、体积分数70%乙醇的洗涤、DNA的溶解及RNA酶酶解等步骤。然后用1%琼脂糖凝胶电泳对提取的基因组进行检测,置于-20℃备用。
1.3.3 PCR扩增及高通量测序
利用引物515F和806R对细菌16SrDNAV4区进行PCR扩增,同时利用引物ITS1F和ITS2对真菌ITS1区进行PCR扩增;最后通过Illumina Miseq测序平台对扩增子片段进行高通量测序。
1.3.4 序列数据处理与分析
得到的高通量测序数据通过去除接头污染、低质量、低复杂度和含N碱基reads获得Clean Data;使用Flash软件利用重叠关系将双末端测序得到的成对reads组成一条序列得到可变区Tags,最后利用UCHIME软件通过与嵌合体数据库比对去除嵌合体得到CleanTags;利用软件USEARCH将Clean Tags进行分类操作单元(operational taxonomic units,OTU)聚类,通过RDP classifer(v2.2)软件将OTU代表序列与数据库比对进行物种注释;最后通过mothur(v1.31.2)软件计算Alpha多样性值并用R(v3.1.1)软件做出相应的稀释曲线图。
2 结果与分析
2.1 酒曲OTU统计
利用软件USEARCH在97%相似度下将Clean Tags聚类为用于物种分类的OTU。OTU可初步说明样品物种丰富程度。经分析,酒曲中的细菌OTU数为47,真菌OTU数为11,说明酒曲微生物组成中细菌种类更为丰富。
根据每个OTU在酒曲样品中的相对丰度,按照丰度从大到小排列,以OTU序号作为横坐标,OTU的相对丰度作为纵坐标使用R(v3.1.1)软件作图,得到OTU Rank曲线(见图1)。根据OTU统计结果和OTU Rank曲线图可知,酒曲中细菌种类较真菌种类多,从每种OTU的相对丰度来看,无论细菌还是真菌在酒曲中物种组成并不均匀,说明酒曲中不同物种含量相差较大。
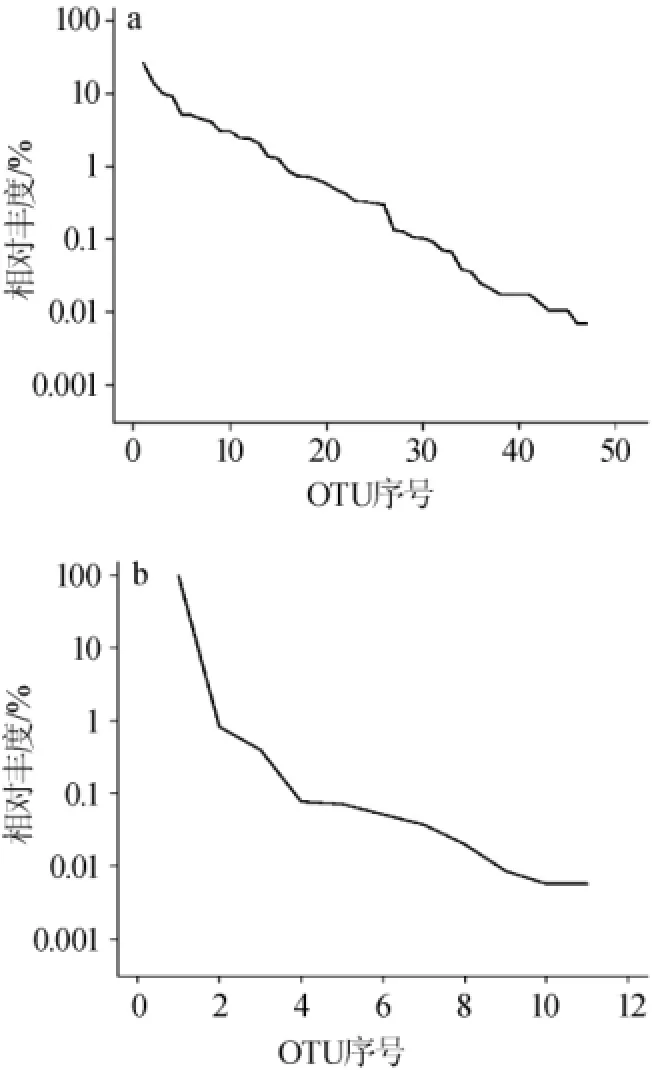
图1 大曲细菌(a)与真菌(b)OTU Rank曲线Fig.1 OTU Rank curve of bacteria(a)and fungi(b)fromDaqu
2.2 酒曲物种及其丰度分析
通过RDP classifer(v2.2)软件对OTU代表序列分别与细菌和真菌数据库比对进行物种注释。对酒曲细菌以及真菌中各物种在门、纲、目、科、属、种6个分类水平上的相对丰度进行统计,结果分别见表1、表2。
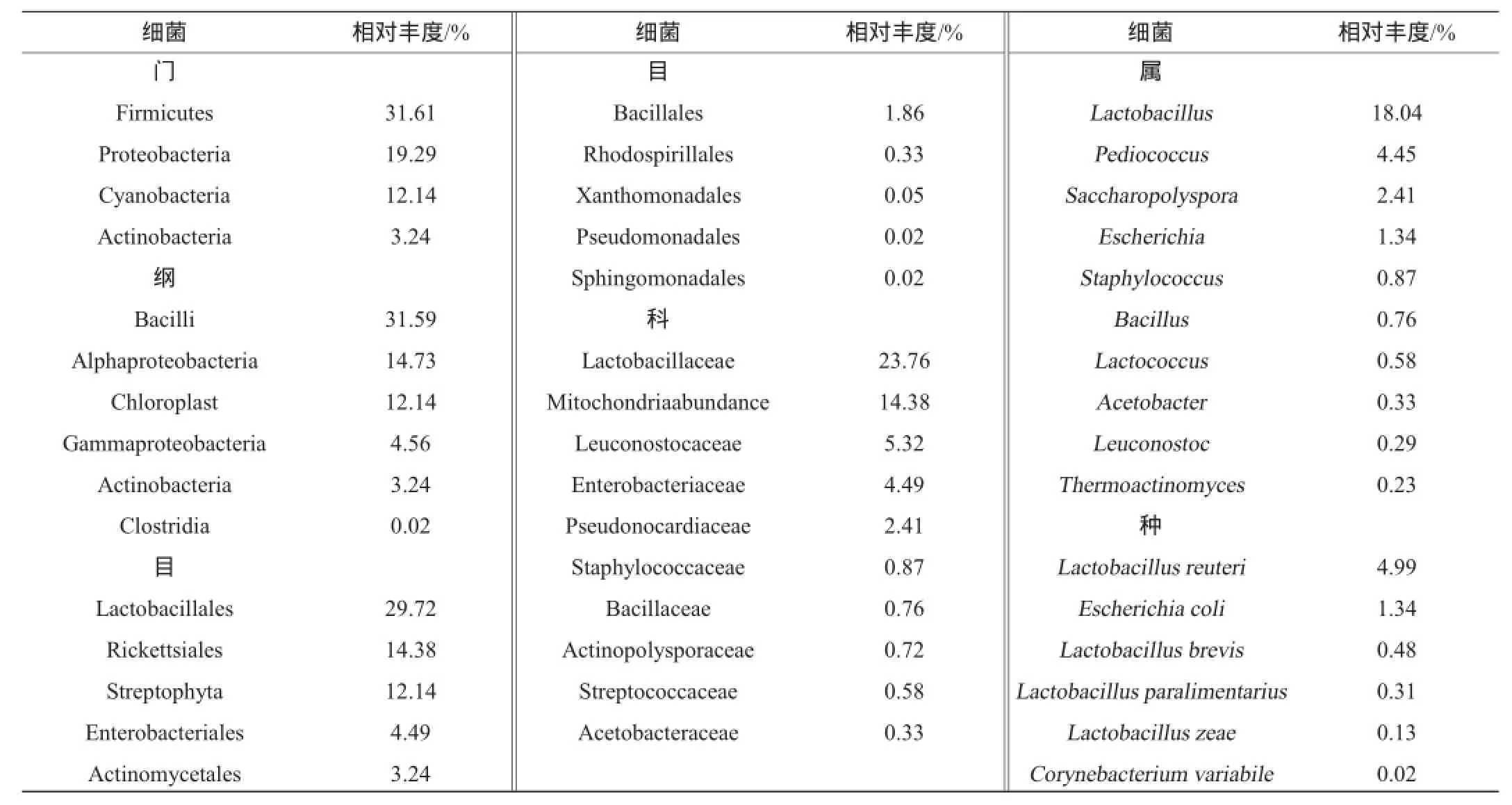
表1 大曲细菌不同分类水平相对丰度统计Table 1 Relative abundance ofDaqubacteria in different classification levels
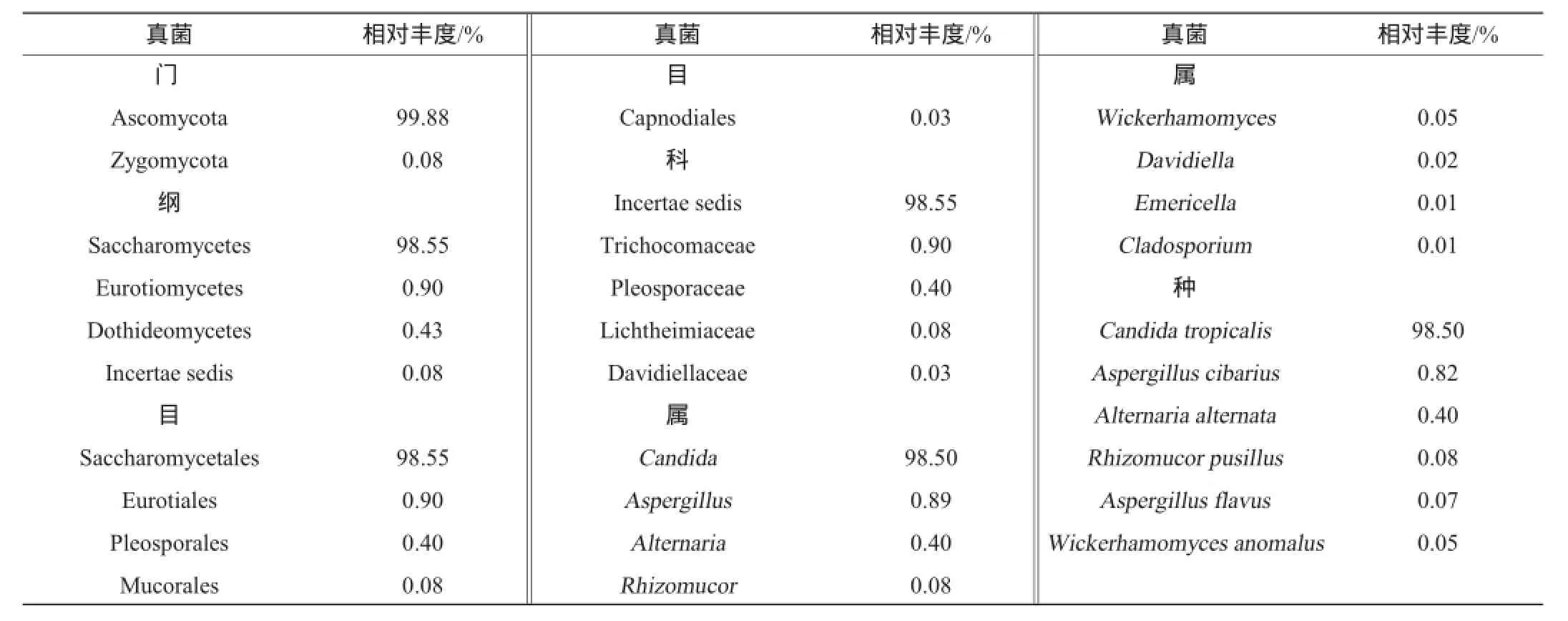
表2 大曲真菌不同分类水平相对丰度统计Table 2 Relative abundance ofDaqufungi in different classification levels
根据酒曲物种相对丰度统计表可知,细菌中以Firmicute(厚壁菌门)下的Bacilli(芽孢杆菌纲)下的Lactobacillales(乳酸杆菌目)为优势菌群,相对丰度值为29.72%;在属分类水平,乳酸杆菌属(Lactobacillus)相对丰度值为18.04%、片球菌属(Pediococcus)相对丰度值为4.45%、乳球菌属(Lactococcus)相对丰度值为0.58%、明串珠菌属(Leuconostoc)相对丰度值0.29%,这些细菌均为乳酸菌,此外,芽孢杆菌属(Bacillus)相对丰度值为0.76%、埃希氏杆菌属(Escherichia)相对丰度值为1.34%、醋杆菌属(Acetobacter)相对丰度值为0.33%、葡萄球菌属(Staphylococcus)相对丰度值为0.87%、高温放线菌属(Thermoactinomyces)相对丰度值为0.23%、糖多孢菌属(Saccharopolyspora)相对丰度值为2.41%,同时一些丰度很低的菌属(表1未列出)也被检测到,如棒状杆菌属(Corynebacterium)相对丰度值为0.02%、黄单胞菌属(Xanthomonas)相对丰度值为0.01%、鞘氨醇单胞菌属(Sphingomonas)相对丰度值为0.02%等,还有许多未分类的物种。
酒曲中真菌种类较少,以热带假丝酵母(Candidatropicalis)为优势菌种,相对丰度高达98.5%;曲霉属(Aspergillus)相对丰度值为0.89%、根毛霉属(Rhizomucor)相对丰度值为0.08%、交链孢属(Alternaria)相对丰度值为0.40%、翘孢霉属(Emericella)相对丰度值为0.01%、威克汉姆酵母属(Wickerhamomyces)相对丰度值为0.05%等均被检测到。
2.3 酒曲样品Alpha多样性分析
Alpha多样性(Alpha diversity)是对单个样品中物种多样性的分析,包括observed species指数、chao指数、ace指数,shannon指数以及simpson指数等。observed species指数、chao指数和ace指数反映样品中群落的丰富度(species richness),这3个指数对应的稀释曲线还可以反映样品测序量是否足够,稀释曲线是从已测得的序列中随机抽取n条序列计算对应的Alpha指数的期望值,然后根据n值与其相对应的Alpha指数的期望值绘制曲线。本次研究分别针酒曲中细菌和真菌用R(V3.1.1)软件作出的Alpha指数稀释曲线图,结果分别见图2、图3。
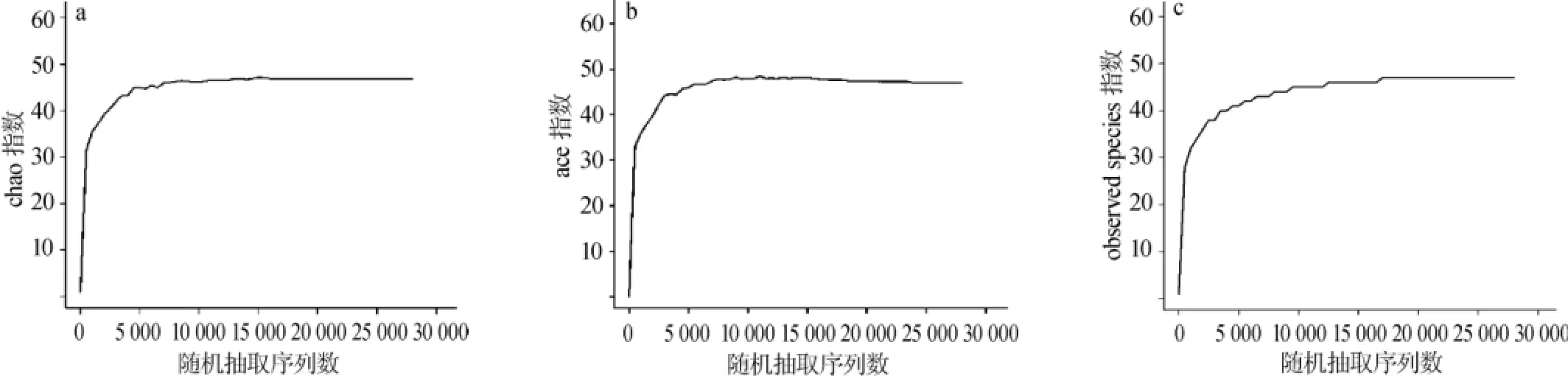
图2 大曲细菌Alpha多样性指数稀释曲线Fig.2 Alpha diversity rarefaction curve ofDaqubacteria
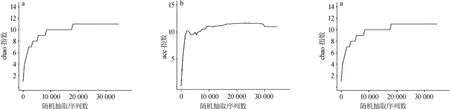
图3 大曲真菌Alpha多样性指数稀释曲线Fig.3 Alpha diversity rarefaction curve ofDaqufungi
由酒曲Alpha多样性指数稀释曲线图可知,随着抽取序列数增大,真菌和细菌稀释曲线均趋于平缓,说明本次测序深度已经基本覆盖到样品中所有的物种。从图3细菌Alpha多样性指数稀释曲线可以看出只要对15 000条序列进行测定分析就可以覆盖酒曲所有细菌,而本次研究细菌多样性分析了28294条序列;同样地由图4可知,只要对20000条序列进行测定分析就能基本覆盖酒曲所有真菌,而实际分析了34 931条序列。因此,本次对酒曲微生物多样性的分析已基本覆盖酒曲中微生物种类。
2.4 讨论
乳酸菌作为大曲中的优势细菌对于白酒中风味物质的产生至关重要。乳酸菌在白酒发酵过程能产生乳酸,乳酸又是形成乳酸乙酯的前体物质,而乳酸乙酯是清香型白酒中除乙酸乙酯外含量最多的酯类物质,适量的乳酸乙酯能增加酒体的醇厚感。乳球菌除发酵产乳酸外,也可生成乙醇、乙酸、甘油等副产物。此外,乳酸菌的代谢产物能够维持白酒发酵的酸性环境[11-12]。除乳酸菌外,酒曲中还有芽孢杆菌、醋杆菌等细菌在白酒酿造过程中发挥重要作用。一般认为芽孢杆菌能产生淀粉酶和蛋白酶分解淀粉和蛋白质等大分子物质[13-14],此外,有些芽孢杆菌能影响白酒风味物质如酯、醇的形成[15]。在白酒生产过程中,醋酸菌的主要代谢产物是醋酸,是白酒中主要的酸类物质,同时也是丁酸、己酸及酯类的前体物质,此外,醋酸菌也可以氧化葡萄糖生成少量酒精[16]。
有很多假丝酵母属菌株都具有酒精发酵能力,有些假丝酵母还具有生香作用[17-18]。曲霉属真菌在白酒发酵过程中可形成糖化力、液化力、蛋白质分解力,降解大分子物质如蛋白质、淀粉形成还原糖、氨基酸、多肽等[19],产风味物质微生物可以进一步利用这些小分子物质生产多种多样的香味物质。
酒曲质量的好坏直接影响白酒的质量和风格。酒曲制作即培养微生物的过程,酒曲为微生物的生长代谢提供了良好的环境,使得不同来源微生物可以共存其中;在制曲过程中,表现出微生物群落交替更迭的现象,形成了非常宝贵的微生物资源库[20]。利用高通量测序技术能够快速便捷地分析酒曲中微生物群落结构,了解酒曲微生物分布及其中的优势菌群,同时能发现一些未知的微生物。
3 结论
本研究基于高通量测序技术在不同分类水平上全面系统地分析了北京清香型酒曲细菌和真菌的多样性,对酒曲样品进行Alpha多样性分析表明本次测序深度足以覆盖其微生物种类。
本次研究结果表明,酒曲中细菌OTU数为47,真菌OTU数为11,且每种OTU在酒曲中分布并不均匀;酒曲中细菌种类比真菌种类丰富;对微生物的相对丰度进行统计表明,乳酸菌是北京清香型酒曲中的一类优势细菌,主要的乳酸菌包括乳酸杆菌属、片球菌属、乳球菌属、明串珠菌属四大类,还有其他细菌如芽孢杆菌、醋酸菌等;假丝酵母属是酒曲中的优势真菌,霉菌也被检测到如曲霉属、根毛霉属等;此外也检测到一些未分类的微生物。微生物研究将会是白酒永恒的课题,分子生物技术的发展必将促进白酒微生物的研究进展,使人们对白酒生产过程中微生物的认识不断深入,从而促进我国白酒的进一步发展。
[1]傅金泉.中国酒曲的起源与发展史探讨[J].中国酿造,2010,29(6):180-186.
[2]荣瑞金,祖明,王德良,等.中国酒曲微生物研究进展[J].中国酿造,2009,28(6):5-8.
[3]敞颜,赵辉,凌宏志.传统白酒发酵微生物的研究进展[J].食品工业科技,2010,31(9):425-427.
[4]WANG H Y,GAO Y B,XU Y,et al.Characterization and comparison of microbial community of different typical Chinese liquorDaqusby PCRDGGE[J].Lett Appl Microbiol,2011,53(2):134-140.
[5]陈玲,袁玉菊,张文学,等.16S rDNA克隆文库法与高通量测序法在浓香型大曲微生物群落结构分析中的对比研究[J].酿酒科技,2015(12):33-36.
[6]秦楠,栗东芳,杨瑞馥.高通量测序技术及其在微生物学研究中的应用[J].微生物学报,2011,51(4):445-457.
[7]王兴春,杨致荣,王敏,等.高通量测序技术及其应用[J].中国生物工程杂志,2012,32(1):109-114.
[8]乔晓梅.清香大曲糖化力酯化力功能及真菌群落结构分析[D].临汾:山西师范大学,2015.
[9]雷振河.采用高通量测序技术分析清香型白酒酿造微生物[J].食品与发酵工业,2015,41(9):164-167.
[10]张瑞福,曹慧,崔中利,等.土壤微生物总DNA的提取和纯化[J].微生物学报,2003,43(2):276-281.
[11]汤洪艳.固态法白酒生产应用乳酸菌的分析[J].中国科技博览,2015(35):373.
[12]孙超,刘勇.白酒生产中乳酸菌的分布及主要代谢产物[J].中国酿造,2012,31(5):1-4.
[13]庄名扬.中国白酒香味物质形成机理及酿酒工艺的调控[J].酿酒,2007,34(2):109-113.
[14]BEAUMONT M.Flavouring composition prepared by fermentation withBacillusspp.[J].Int J Food Microbiol,2002,75(3):189-196.
[15]马明,杜金华.枯草芽孢杆菌在工业生产中的应用[J].山东科学,2006,19(3):35-38.
[16]侯小歌,杜红阳,胡炳义,等.宋河大曲中醋酸菌的分离鉴定及产酸特性[J].中国酿造,2011,30(4):112-115.
[17]沈怡方.白酒生产技术全书[M].北京:中国轻工业出版社,1998,48.
[18]WANG C L,SHI D J,GONG G L.Microorganisms inDaqu:a starter culture ofChinese Maotai-flavor liquor[J].World J Microbiol Biotechnol,2008,24:2183-2190.
[19]KEN O,KAZUHIRO I,DARARAT K,et al.Proteomic analysis of extracellular proteins fromAspergillus oryzaegrown under submerged and solid-state culture conditions[J].Appl Environ Microbiol,2006, 72(5):3448-3457.
[20]申孟林,张超,王玉霞.白酒大曲微生物研究进展[J].中国酿造,2016,35(5):1-5.
Analysis on microbial diversity of Beijing light-flavorDaquby high-throughput sequencing
ZHANG Shuangyan1,LIAO Yonghong2,JI Nan2,ZHOU Xiaohong1*
(1.School of Life Science,Beijing Institute of Technology,Beijing 100081,China;2.School of Food and Chemical Engineering, Beijing Technology and Business University,Beijing 100048,China)
In order to analyze microbial diversity in Beijing light-flavorDaqu,16S rDNA V4 region and ITS1 region were analyzed by high-throughput sequencing technology,then the relative abundance ofDaqumicrobes in the different classification levels were counted.The results showed that Daqubacteria operational taxonomic units(OTU)was 47,fungi OTU was 11,the varieties of bacteria was more abundant than that of fungi.Lactic acid bacteria were the predominant bacteria,includingLactobacillus,Pediococcus,Lactococcus,Leuconostoc,andBacillusandAcetobacterwere also detected.Candidawas the predominant fungi.Aspergillus,Rhizomucor,Wickerhamomycesand other genus were also identified.Alpha diversity analysis showed that the number of analyzed sequences was enough to cover microbial species inDaqu.Finally,the possible function of dominant microbes in the process ofBaijiu-making was discussed.This study had important significance for the guidance ofBaijiu(Chinese liquor)production.
light-flavorDaqu;high-throughput sequencing;microbe;16S rDNA V4;ITS
Q93-3;TS261.1;TQ920
0254-5071(2016)11-0049-05
10.11882/j.issn.0254-5071.2016.11.010
2016-08-18
国家“十二五”科技支撑计划项目(2014BAC28B01)
张双燕(1992-),女,硕士研究生,研究方向为食品微生物。
*通讯作者:周晓宏(1965-),男,副教授,博士,研究方向为食品生物技术。
