含N-非取代葡糖胺残基肝素四糖的制备与表征
2016-12-10梁群焘
梁群焘,魏 峥
(福州大学糖生物化学研究所,福建 福州 350002)
含N-非取代葡糖胺残基肝素四糖的制备与表征
梁群焘,魏 峥
(福州大学糖生物化学研究所,福建 福州 350002)
生物体内含有N-非取代葡糖胺残基(GlcNH3+)结构的硫酸类肝素(HS)具有重要的生物和病理生理学功能。但这种HS在生物体内的含量较少、获得困难,而采用化学方法制备与生物体内结构相似的这种寡糖,有助于研究HS在生物体内的功能作用。本实验以高硫的肝素四糖为原料,用部分脱N位硫酸根的方法,制备了含1个和2个GlcNH3+的肝素四糖,并采用液相色谱-离子阱-飞行时间质谱(LC/MS-IT-TOF)法对其进行结构检测。通过分析(EIC)-MS和MS2提取离子流图发现,含不同GlcNH3+数目的肝素四糖具有不同的裂解规律,含GlcNH3+数目越多,生成的碎片离子越多,这为MS方法进一步鉴定和定量测定含GlcNH3+结构的寡糖奠定了基础。
肝素;四糖;N-非取代葡糖胺二糖;液相色谱-离子阱-飞行时间质谱(LC/MS-IT-TOF);裂解规律
肝素(heparin)和硫酸类肝素(HS)在生物个体发育,血管生成,血液凝固,细胞粘附和肿瘤转移等过程中具有重要的生物学活性作用[1-4]。其生物学功能与它们的结构多样性,以及与细胞表面和细胞外蛋白相互的作用密切相关,且糖胺聚糖(GAG)的结构决定了它们相互作用的能力[5-6]。
在高尔基体中,肝素和硫酸类肝素的生物合成是通过多种酶间复杂的相互协同作用完成的,其原理示于图1[7]。首先,形成1个葡萄糖醛酸-半乳糖-半乳糖-木糖(glucuronic acid-galactose-galactose-xylose,GlcUAβ1→3Galβ1→3Galβ1→4Xylβ1-)的“连接四糖”结构,把HS链连接到蛋白聚糖核心蛋白的丝氨酸上;接着,通过复杂的EXT1和EXT2聚合酶催化交替转移葡萄糖醛酸(GlcUA)和N-乙酰葡糖胺(GlcNAc),合成非硫酸化的HS初生链。HS初生链由重复的二糖组成,这些二糖由β1→4连接GlcUA和α1→4连接GlcNAc相互交替组成。在N-脱乙酰/N-磺基转移酶(N-deacetylase/N-sulfotransferase,NDST)的作用下,非硫酸化的初生链GlcNAc基团在N位上脱乙酰化形成1个N-非取代葡萄糖胺(GlcNH3+)中间体结构,随后进一步的修饰[8-11]。如,GlcUA残基C-5差向异构化(C5-epimerase)为α1→4艾杜糖醛酸残基(IdoA);在HS 3-O-磺基转移酶(3-O-sulfotransferases,3-OST)的作用下,GlcNS的C-3位氧硫酸化,形成一种比较罕见但功能重要的残基;己糖醛酸(主要是IdoA)C-2位氧硫酸化;GlcNS或GlcNAc残基上的C-6位氧硫酸化;N位硫酸化,游离氨基基团被硫酸化取代形成带负电荷的氨基硫酸衍生物,即β-D-N-硫葡糖胺硫酰胺(GlcNS)。在肝素和硫酸类肝素生物合成过程中,由于NDST酶活性受到限制,N-乙酰葡糖胺(GlcNAc)残基上产生随机的N-脱乙酰化,但不发生随后的N-磺酸化过程,从而产生了GlcNH3+。这种现象在缺少3′-磷酸腺苷-5′-磷酸硫酸酯(PAPS)的情况下更为明显。这是因为PAPS是硫酸盐供体,缺乏PAPS可导致NDST酶的催化活性受到限制[12]。
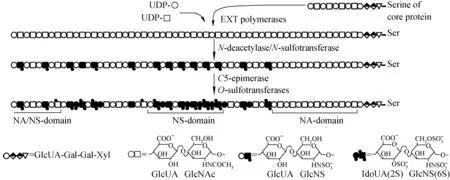
图1 硫酸类肝素的生物合成原理示意图[7]
生物体内HS的GlcNH3+含量较低,且因物种、组织和细胞的来源不同而异,一般含GlcNH3+的二糖含量为0.2%~4%[13-17],但在牛肾HS中,GlcNH3+的含量高达12%[16]。但低含量的GlcNH3+残基却与许多重要的细胞生物活动和病理生理现象有关[18-20]。硫酸化的二糖结构IdoA(2S)-GlcNH3+(±6S)是HS 3-O磺基转移酶-3(3-OST-3)的作用靶点,这种HS酶能够生成一个位点,这个位点可以与单纯疱疹病毒HSV[19]以及人嗜环蛋白/亲环素B(CyPB)的gD糖蛋白[21]产生粘附结合作用,并且这种硫酸化的二糖结构主要位于HS的高硫区域。化学合成的含GlcNH3+残基的HS四糖GlcAβ1-4 GlcNH3+(6-O-sulfate)α1-4 GlcAβ1-4 GlcNH3+(6-O-sulfate),在体外能够抑制肝素酶的活性,并抵抗乳腺癌细胞的侵袭[22]。鉴于含GlcNH3+的HS/heparin寡糖具有重要的生物学功能,但其含量较少,难于获得,因此制备含GlcNH3+且与生物体内结构类似的高度硫酸化HS/heparin寡糖,对于研究含GlcNH3+的HS/heparin有关生物学功能具有重要意义。目前,HS/heparin寡糖的制备多集中在常用的肝素类药物上,而含少量GlcNH3+残基的HS/heparin寡糖制备的报道较少。如,Satomi等[22]在体外合成了一种含GlcNH3+的HS四糖,并研究其功能;Wei等[23]用肝素酶Ⅰ裂解的方法制备了系列含GlcNH3+残基的HS/heparin寡糖,但酶解产率较低。
质谱具有检测灵敏度高,能够准确测定物质的分子质量等优点,现已成为分析HS/heparin寡糖结构的有力工具[24-26]。采用MSn分析heparin和HS,能获得丰富的糖苷键断裂以及环内裂解的碎片信息,这些信息可用来确定硫酸化和乙酰化的作用位点,并依此判断物质的结构[27]。有报道采用ESI-MSn方法在负离子模式下探析12种硫酸类肝素二糖[28]和8种CS/DS二糖[29]的裂解规律,特别是利用计算框架处理MS生成的复杂数据,使得LC/MS方法具有检测大多数寡糖序列的潜力[30]。文献[31]报道了一种新的含GlcNH3+寡糖的测序方法,它结合了HNO2裂解、尺寸排阻高效液相色谱(SE-HPLC)和液相色谱-离子阱-飞行时间质谱(LC/MS-IT-TOF)方法,为确定化学修饰生成的寡糖结构系列提供了可能。
本实验将制备含不同GlcNH3+数目的肝素四糖(dp4),采用LC/MS-IT-TOF法对其进行结构分析,并探讨其在MS分析中的裂解规律,希望为含GlcNH3+寡糖的结构分析提供方法参考,同时也为检测含GlcNH3+寡糖的生物样品提供技术支持。
1 实验部分
1.1 主要仪器
高效液相色谱-离子阱-飞行时间质谱仪(LC/MS-IT-TOF MS):日本岛津公司产品,配有二元泵(LC-20AD)、脱气装置(DGU-20A3R)、自动进样系统(SIL-20AC)、光敏二极管阵列(PDA)、检测器(SPD-M20A)、基本通信模块(CBM-20A)和柱温箱(CTO-20A);1200型高效液相色谱仪:美国Agilent公司产品;Milli-Q超纯水机:美国Millipore公司产品;Alpha 1-4 LDplus型真空冷冻干燥机、CT02-50SR型恒温真空冷冻浓缩仪:均为德国Christ公司产品。
1.2 主要材料与试剂
低分子质量肝素(LMWH, Innohep):英国Leo制药有限公司产品;12种硫酸类肝素二糖标样:英国Iduron公司产品;Bio-Gel P-10聚丙烯酰胺凝胶:英国Bio-Rad公司产品;MWCO 500透析膜:上海索莱宝生物技术有限公司产品;肝素酶Ⅰ(50 UN)、肝素酶Ⅱ(10 UN)、离子交换树脂IR-120(H+型)、吡啶(纯度>99.9%)、二甲亚砜(分析纯)、碳酸氢铵(分析纯)、氯化钠(纯度≥98%)、盐酸(分析纯)、甲酸(纯度98%)、戊胺(纯度99%):均为美国Sigma-Aldrich公司产品;乙腈(色谱纯):德国Merck公司产品。
1.3 实验方法
1.3.1 高度硫酸化肝素四糖的制备 高度硫
酸化肝素四糖(dp4)的制备参考文献[23],主要由两个步骤组成。步骤一:用Bio-Gel P-10凝胶分析分离LMWH,以0.2 mol/L NH4HCO3为流动相,UV 232 nm检测洗脱液吸光度,收集肝素dp4混合物的洗脱峰;然后在50~60 ℃水浴中恒定72 h,去除NH4HCO3,于-80 ℃冷冻5 h,真空干燥,得到肝素dp4混合物样品。步骤二:用强阴离子交换高效液相色谱(SAX-HPLC)分析分离肝素dp4混合物,收集高硫dp4部分;然后用透析膜在超纯水中透析3天,去除NaCl,于-80 ℃冷冻5 h,真空干燥,得高度硫酸化的肝素dp4样品。1.3.2 含GlcNH3+肝素四糖的制备 含GlcNH3+肝素四糖是通过高度硫酸化dp4部分脱N位硫酸根获得的,其方法参考文献[23],过程示于图2,主要由三个步骤组成。步骤一:用超纯水溶解高硫dp4,在4 ℃下缓慢经过H+型离子交换树脂,去除高硫dp4上的Na+;然后用3倍树脂体积的4 ℃超纯水洗脱。步骤二:用吡啶滴定洗脱液至pH 6~6.5,获得以吡啶盐形式存在的dp4。步骤三:在95%二甲亚砜-5%水溶液(V/V)、18~20 ℃条件下,吡啶盐dp4脱N位硫酸根15 min;然后用透析膜在超纯水中透析纯化3天,于-80 ℃冷冻5 h,真空干燥,得含N-非取代葡糖胺残基的dp4。

图2 肝素四糖部分脱N位硫酸根步骤示意图
1.3.3 肝素寡糖二糖组分分析 在100 μL 0.1 mol/L醋酸钠缓冲液(pH 7.0),含0.1 mmol/L醋酸钙和100 mg/L牛血清蛋白反应体系下,用肝素酶Ⅰ(50 UN)和肝素酶Ⅱ(0.5 UN)裂解肝素寡糖,于37 ℃反应24 h,100 ℃灭活2 min,以15 000 r/min离心15 min,取上清液。酶解产物用SAX-HPLC分析,通过比对检测图谱和标样二糖图谱来确定二糖组分。
1.3.4 SAX-HPLC分析 ProPac PA-1色谱柱(4.0 mm×250 mm);洗脱程序:用pH 3.5的H2O冲洗2 min后,在0~0.6 mol/L NaCl(2.1~7.1 min)和0.6~1.3 mol/L NaCl(7.1~47.1 min)条件下,两相线性梯度洗脱;流动相:pH 3.5的H2O(A相),pH 3.5的2 mol/L NaCl(B相);流速1 mL/min;柱温:室温;在线检测器:UV232nm。
1.3.5 LC/MS-IT-TOF分析 色谱条件:Eclipse Plus C18色谱柱(3.5 μm×2.1 mm×150 mm);流速0.2 mL/min;柱温35 ℃;流动相为H2O(A相)和75%乙腈(B相),含有15 mmol/L戊胺(PTA)离子对试剂,用甲酸调至pH 8.8;洗脱程序为5%~100%乙腈(2~20 min)线性梯度洗脱;光敏二极管阵列检测范围为190~800 nm。
质谱条件:电喷雾离子源(ESI),负离子模式;质量扫描范围m/z200~1 800;喷雾气体为液氮,流速1.5 L/min;曲形脱溶剂管和加热模块温度设置为100 ℃;锥孔电压-3.5 kV;检测电压1.6 kV;IT和TOF真空分别保持在1.8×10-2Pa 和1.6×10-4Pa;质量轴校准采用直接注入标准样品(0.25 mL/L三氟乙酸和0.1 g/L NaOH的混合物)的方法,检测范围m/z150~2 100,流速0.2 mL/min,经过质量校准的所有质量数参考标准偏差不超过5×10-6。
1.3.6 MS/MS条件 样品溶解于10%乙腈,以0.2 mL/min流速直接注入LC/MS-IT-TOF中;设置质谱系统为自动模式,自动选择强度高于105的产物离子作为MS2分析的母离子;碰撞气体为氩气;离子累积时间设为100 ms;其他质谱条件同1.3.5节。
2 结果与讨论
2.1 高度硫酸化肝素四糖的分析
LMWH是肝素酶Ⅰ的酶解产物,在己糖醛酸非还原端 C4和C5位置具有1个不饱和双键。以0.2 mol/L NH3HCO3为流动相,Bio-Gel P-10可以把不同聚合度的肝素寡糖分开,得到的dp4用ProPac柱和SAX-HPLC方法进一步分离纯化,结果示于图3。结果表明,dp4有3个主要的峰,根据出峰时间分别命名为dp4A、dp4B 和dp4C。按电荷密度不同进行分离,dp4C具有最长的保留时间,说明它含有最多的硫酸基团。二糖组分分析表明,dp4C只含有ΔHexA(2S)-GlcNS(6S),说明dp4C含有2个高硫的二糖,即ΔHexA(2S)-GlcNS(6S)-HexA(2S)-GlcNS(6S);结合RPIP-LC/MS分析,可推测dp4C主要成分为含有6个硫酸基团高度硫酸化的dp4。
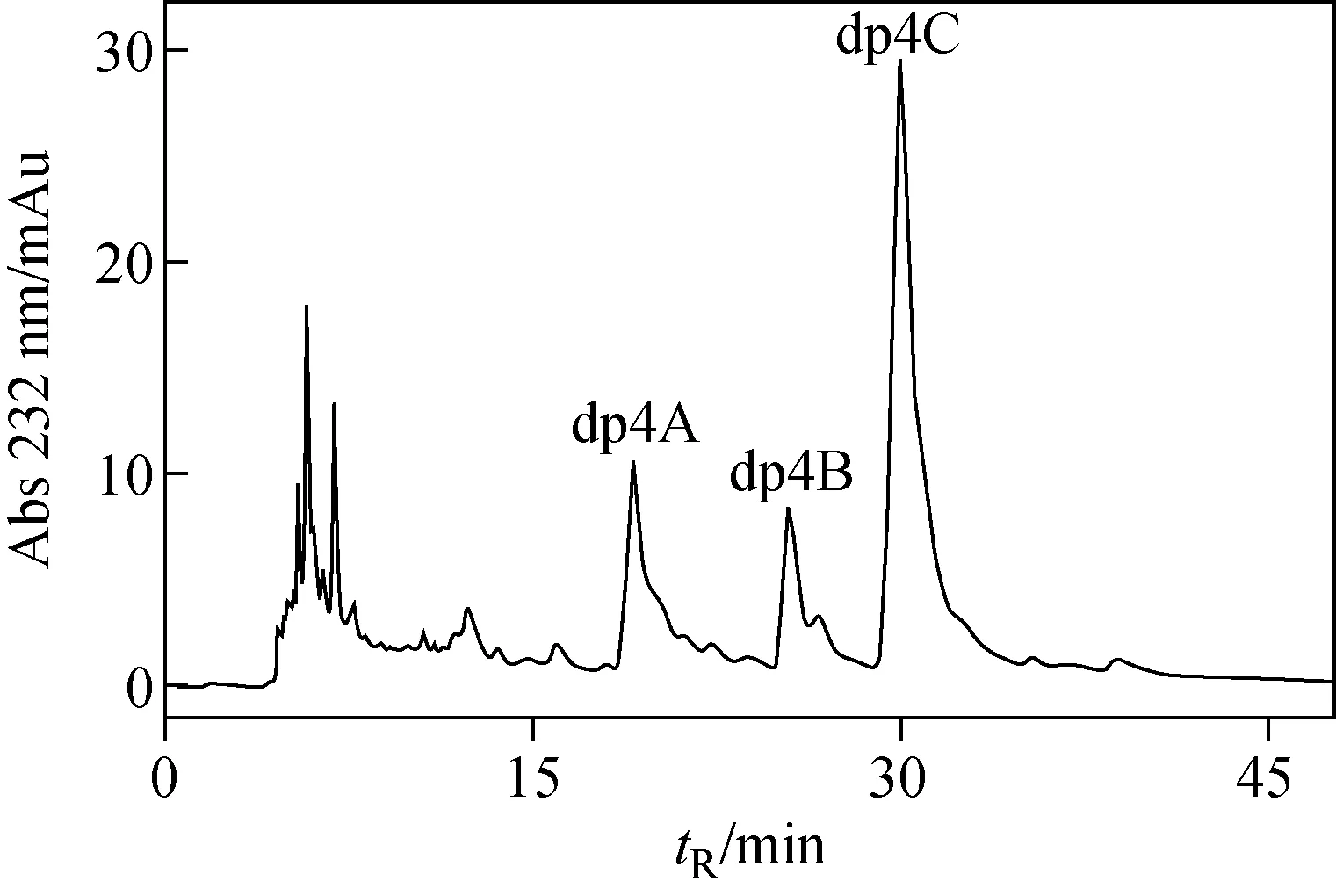
图3 经Bio Gel P-10分离后的肝素四糖的SAX-HPLC分离色谱图
2.2 含N-非取代葡糖胺(GlcNH3+)残基肝素四糖的分析
高度硫酸化的dp4用部分脱N位硫酸根法制得GlcNH3+后,产物经SAX-HPLC分析,出现2个主要的峰,分别命名为dp4-1和dp4-2,示于图4。二糖组分分析表明,dp4-2只含有ΔHexA(2S)-GlcNH3+(6S),说明dp4-2含有2个GlcNH3+残基,即ΔHexA(2S)-GlcNH3+(6S)-HexA(2S)-GlcNH3+(6S);而dp4-1含有ΔHexA(2S)-GlcNH3+(6S)和ΔHexA(2S)-GlcNS(6S) 两种二糖,根据文献[23],确定dp4-1的结构为ΔHexA(2S)-GlcNH3+(6S)-IdoA(2S)-GlcNS(6S)。研究heparins链上的GlcNH3+基团是制药领域的热点。Heparin在高温或高压蒸气灭菌处理时产生GlcNH3+残基,这会改变高度特异的内源性抗凝血酶Ⅲ结合位点,导致heparin抗凝能力下降[32-33],而化学合成的含GlcNH3+残基的HS四糖在体外能抑制肝素酶的活性、抵抗乳腺癌细胞的侵袭[22]。因此,本实验制备了含GlcNH3+的HS/heparin寡糖,并分别单独收集含不同GlcNH3+数目的dp4-1和dp4-2,透析纯化后进行LC/MS-IT-TOF分析。
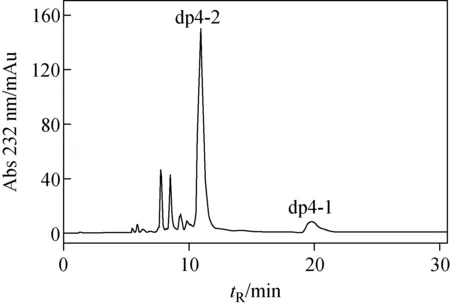
图4 含GlcNH3+肝素四糖的SAX-HPLC色谱图
2.3 RPIP-LC/MS-IT-TOF分析
RPIP-LC/MS法可以有效地分析分离肝素寡糖,且能减少糖链上硫酸根的丢失[34],准确地测量寡糖的分子质量,从而确认寡糖含有的GlcNH3+数目。由于硫酸根比较脆弱,在MS分析过程中容易丢失,因此,本实验采用高硫的dp4C对使用的缓冲体系和MS仪器参数进行优化。采用MS质量数和提取离子流(EIC)对样品进行表征,且用它们各自对应的EIC峰面积计算各离子峰的相对百分含量。dp4的MS谱图示于图5,图中离子峰对应的分子离子表达式列于表1。
dp4C的MS图示于图5a。图中,m/z575.916 7为dp4C带2个负电荷的分子离子峰[M—2H]2-。m/z619.466 8、663.014 6、706.563 5、750.112 7分别为dp4C加合了系列PTA(从1个到4个PTA),且带2个负电荷的分子离子峰,它们大约占总离子峰含量的76%,说明在MS分析过程中,绝大部分的dp4C没有发生硫酸根的丢失。另外,检测到4个dp4C丢失硫酸根的分子离子峰,即[M—4SO3—2H]2-、[M—3SO3—2H]2-、[M—2SO3—2H]2-和[M—SO3—2H]2-,这些丢失硫酸根的分子离子峰与加合PTA的峰具有相同的洗脱位置,说明硫酸根的丢失发生在MS离子源中。
采用dp4C的MS条件分析含有1个GlcNH3+的dp4-1,结果示于图5b。强度最大的离子峰m/z535.942 2为dp4-1带2个负电荷的分子离子峰[M—2H]2-,另外也检测到了dp4-1分别加合1个和2个PTA的分子离子峰,这3种峰大约占总离子峰含量的80%。与dp4C检测结果相似,dp4-1也检测到了3个在MS离子源分别丢失1~3个硫酸根的分子离子峰。
采用同样的MS条件分析含有2个GlcNH3+的dp4-2,结果表明,强度最大的离子峰m/z495.966 7为dp4-2带2个负电荷的分子离子峰[M—2H]2-,且dp4-2大约有相对百分含量为88%的分子离子没有丢失硫酸根,示于图5c。以上数据表明,虽然样品在RPIP-LC/MS-IT-TOF分析过程中有硫酸根丢失的现象,但绝大多数样品保持了最初的结构形态,这为准确测定肝素寡糖的分子质量,确认寡糖含有的GlcNH3+数目提供了帮助。
2.4 肝素dp4s的裂解规律
串联质谱法(MSn)常被用来分析GAGs结构,它能提供丰富的糖苷键断裂以及环内裂解的碎片信息,这些信息可以确定硫酸化和乙酰化的作用位点[27]。本实验采用MS/MS法探讨dp4C、dp4-1和dp4-2的裂解规律,并分析它们的结构,结果示于图6~8。MS/MS产物离子的表述采用Domon和Costello命名规则[35]。
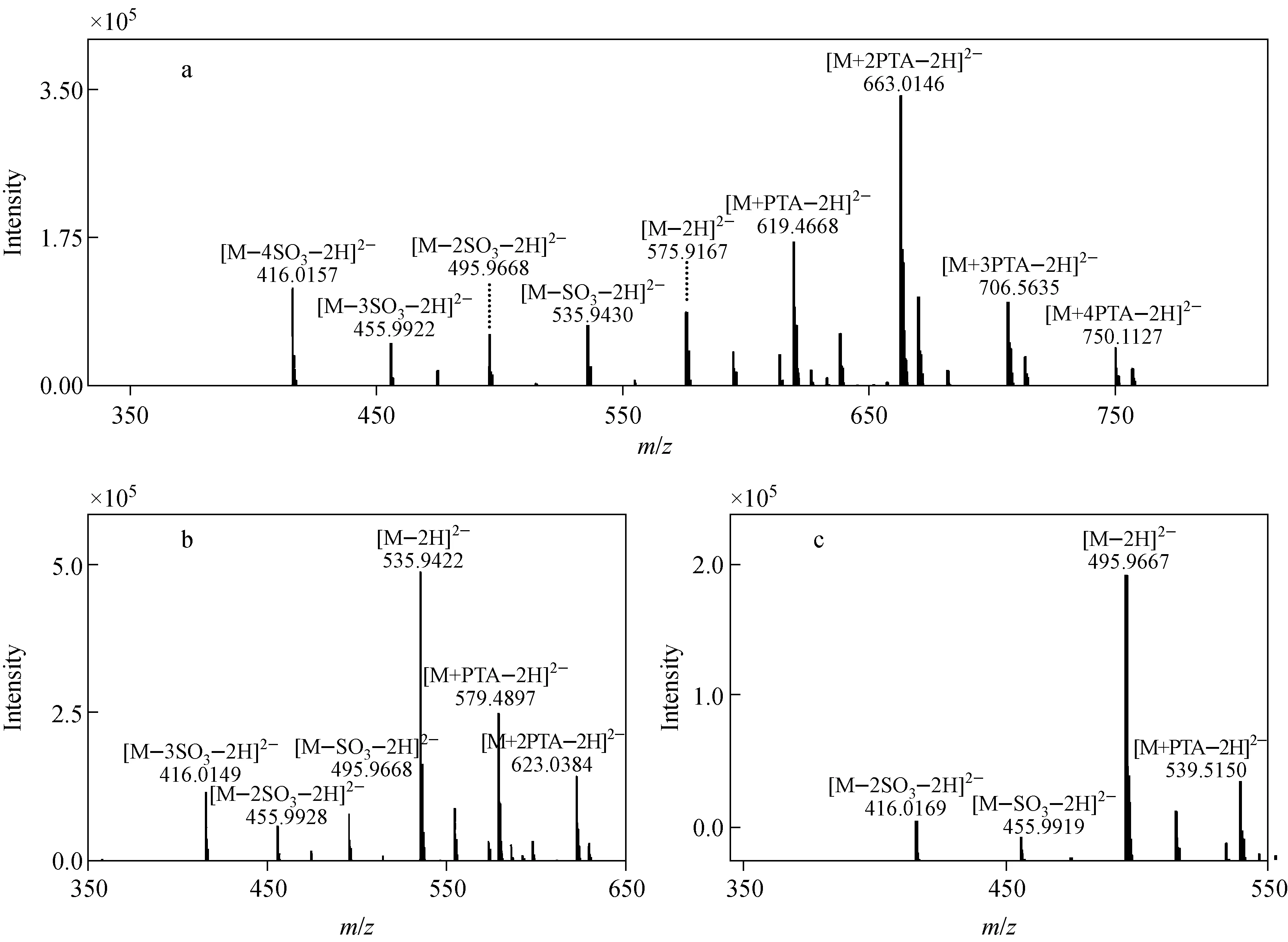
注:a.高硫dp4C;b.含1个残基的dp4-1;c.含2个残基的dp4-2
表1 高度硫酸化的dp4C、含1个GlcNH3+的dp4-1、含2个GlcNH3+的dp4-2在ESI-MS图谱中出现的主要分子离子峰
Table 1 Major molecular ions that appeared in the ESI mass spectra of the fully sulfated dp4C,dp4-1 with one GlcNH3+residue and dp4-2 with two GlcNH3+residues
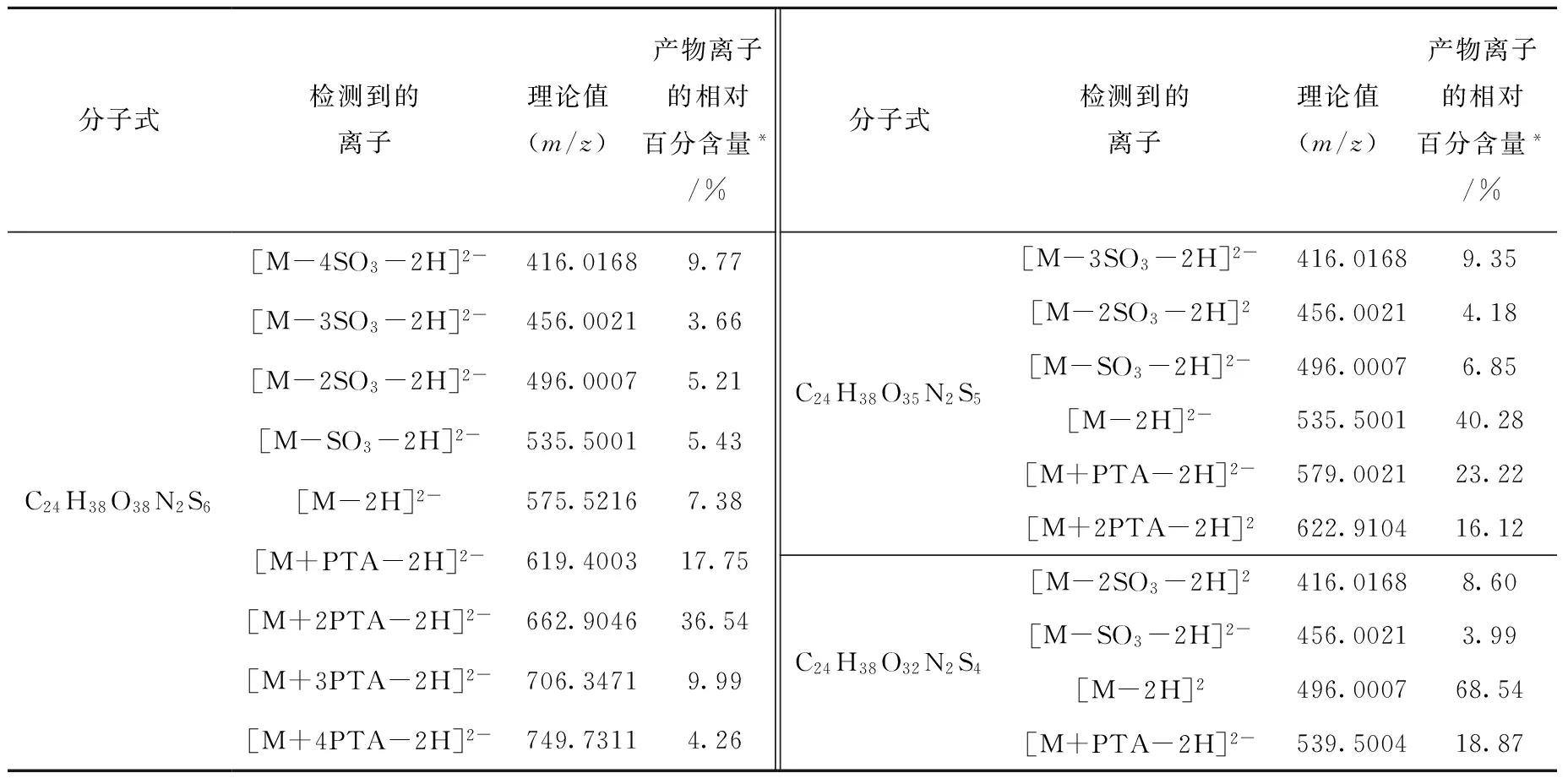
分子式检测到的离子理论值(m/z)产物离子的相对百分含量*/%分子式检测到的离子理论值(m/z)产物离子的相对百分含量*/%C24H38O38N2S6[M-4SO3-2H]2-416.01689.77[M-3SO3-2H]2-456.00213.66[M-2SO3-2H]2-496.00075.21[M-SO3-2H]2-535.50015.43[M-2H]2-575.52167.38[M+PTA-2H]2-619.400317.75[M+2PTA-2H]2-662.904636.54[M+3PTA-2H]2-706.34719.99[M+4PTA-2H]2-749.73114.26C24H38O35N2S5C24H38O32N2S4[M-3SO3-2H]2-416.01689.35[M-2SO3-2H]2456.00214.18[M-SO3-2H]2-496.00076.85[M-2H]2-535.500140.28[M+PTA-2H]2-579.002123.22[M+2PTA-2H]2622.910416.12[M-2SO3-2H]2416.01688.60[M-SO3-2H]2-456.00213.99[M-2H]2496.000768.54[M+PTA-2H]2-539.500418.87
注:*根据它们对应的EIC面积计算
dp4C的ESI-MS结果示于图6a,从图中可以观察到带2个负电荷和带3个负电荷的离子,且带3个负电荷的离子峰强度最大。产物离子大多是带电的分子离子和小分子中性丢失(如SO3、H2O、CH2O 和 CO2H)产生的碎片离子。同时,检测到了糖苷键裂解和环内裂解生成的碎片离子峰,但峰强度较低,如2个带负电荷的离子(m/z284.002 2、m/z407.039 4)分别与碎片离子[Z3—COOH—2H]3-(计算值m/z283.988 4)和[Y3—SO3—H2O—H]2-(计算值m/z407.077 6)对应,说明在MS分析过程中dp4C产生了Z3和Y3糖苷键的裂解;另外,还检测到了2个带负电荷的离子(m/z287.476 9、m/z324.330 0)分别与碎片离子[2,4A3—COOH—H]2-(计算值m/z287.499 0)和[0,2X3—H2O—COOH—H]3-(计算值m/z324.318 33)对应,说明在MS分析过程中dp4C发生了2,4A3和0,2X3环内裂解。
dp4C代表性的母离子[M—2SO3—2H]2-(m/z496.005 6,计算值m/z495.971 0)的ESI-MS2分析结果示于图6b。为了进一步探讨dp4C在MS分析中的裂解规律,ESI-MS产物离子峰强度大于105的离子自动被选为母离子做ESI-MS2分析。结果表明,不同母离子产生的碎片离子数目不同,但检测到的离子m/z数值和带电情况都相似。检测到的m/z307.520 1与碎片离子[2,4X2—3SO3—2H]2-(计算值m/z307.482 65)对应,m/z386.529 1与碎片离子[0,2A4—3SO3—2H]2-(计算值m/z386.475 55)对应,这说明在ESI-MS2中dp4C发生了环内2,4X2和0,2A4裂解。
ESI-MS和MS2检测到的dp4C可能的裂解方式示于图6c。其中,ESI-MS检测到2种糖苷键裂解(Z3、Y3)和2种环内裂解(2,4A3和0,2X3);MS2也检测到Z3、Y3裂解,以及2,4X2和0,2A4环内裂解。
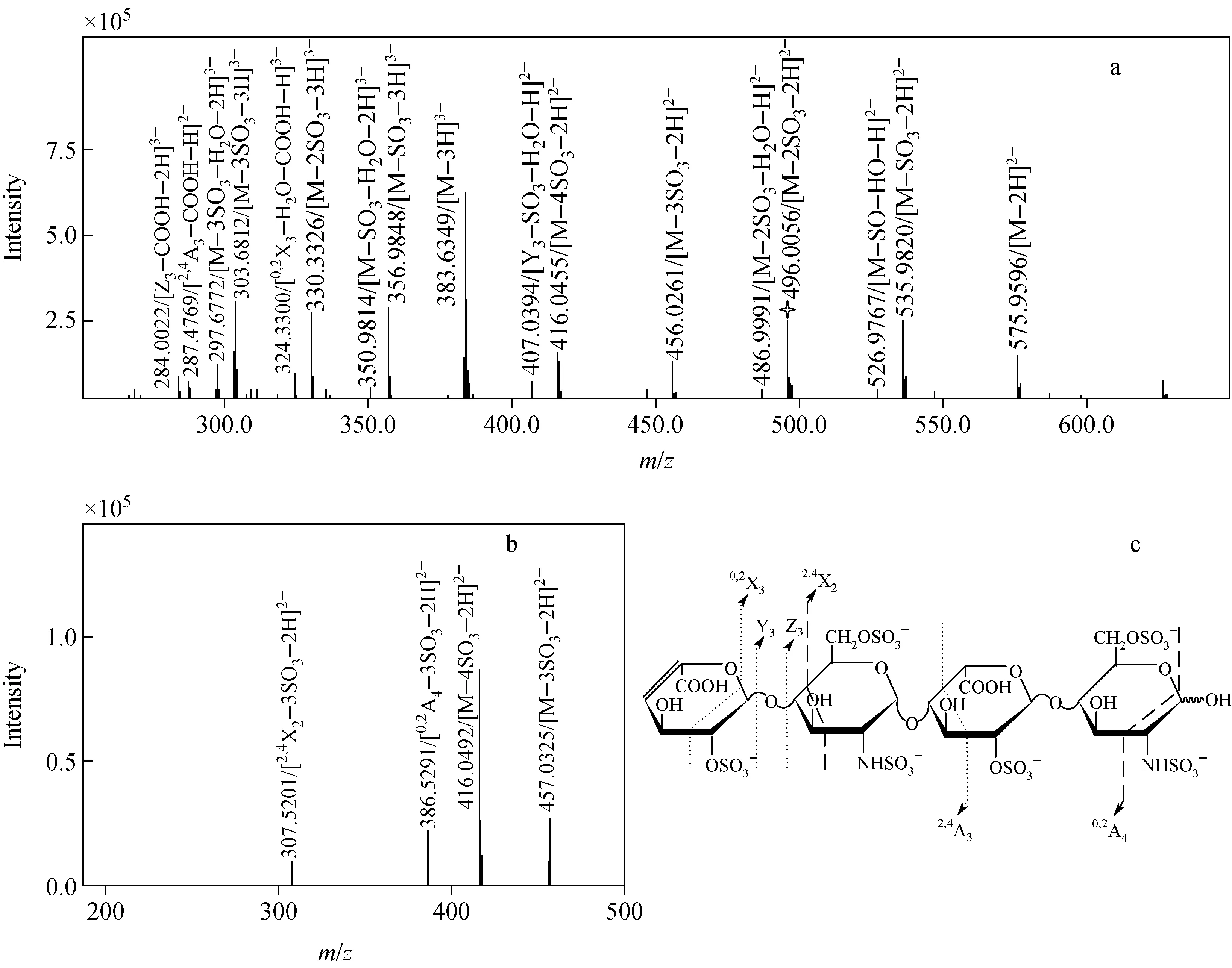
注:a.ESI-MS图谱;b.图a中m/z 496.005 6作为母离子的MS2图谱;
dp4-1的ESI-MS结果示于图7a,同样也检测到带2个和3个负电荷的离子,但带2个负电荷的离子峰强度最大。产物离子大多也是带电的分子离子和小分子中性丢失(如SO3、H2O、CH2O 和 CO2H)产生的碎片离子,糖苷键裂解和环内裂解生成的碎片离子峰强度也较低。
dp4-1代表性的母离子[M—2SO3—2H]2-(m/z456.032 3,计算值m/z456.066 9)的ESI-MS2分析结果示于图7b。同样,为了进一步探讨dp4-1在MS分析中的裂解规律,ESI-MS产
物离子峰强度大于105的离子自动被选为母离子做ESI-MS2分析。dp4-1检测到了特有的裂解方式0,2X2(检测值m/z240.023 1,碎片离子[0,2X2—CH2OSO3—COOH—H]2-,计算值m/z239.982 65)。
ESI-MS和MS2分析中dp4-1的裂解方式示于图7c。检测到5种糖苷键裂解(Y3、Z3、Z2、B3和C3)和6种环内裂解(1,5X3、2,4X2、0,2X2、0,2A3、3,5A4和0,2A4),其中,Y3、3,5A4和0,2A4为ESI-MS检测到的裂解,其余的裂解均发生在ESI-MS2过程中。与dp4C相比,均出现了相同的糖苷键裂解(Y3和Z3)和相同的环内裂解(2,4X2和0,2A4),但0,2X2为dp4-1特有的裂解方式,位于靠近非还原端的含有GlcNH3+基团的葡萄糖胺上。
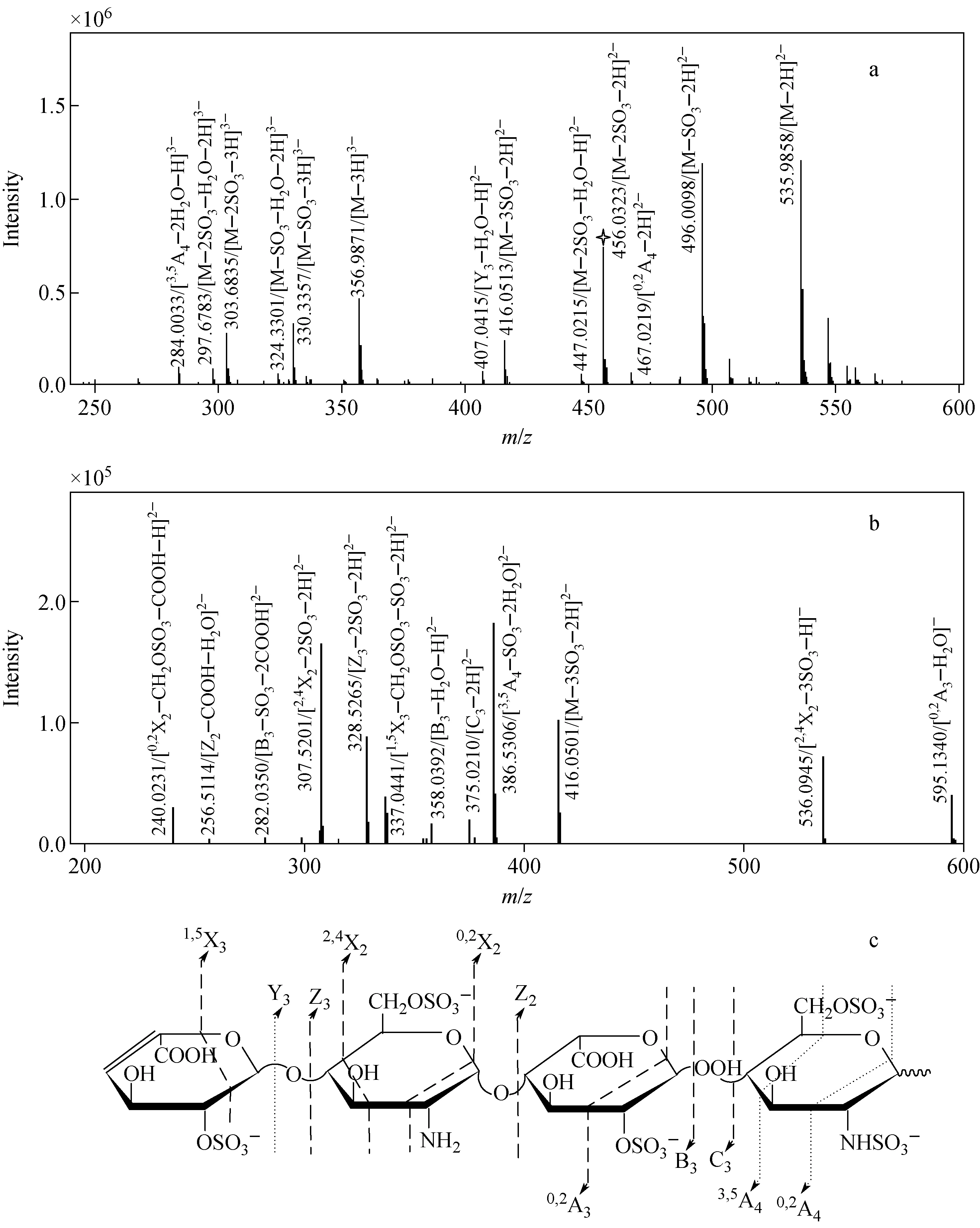
注:a.ESI-MS图谱; b.图a中m/z 456.032 3离子作为母离子的MS2图谱;
在ESI-MS和MS2分析过程中,含2个GlcNH3+基团的dp4-2检测到了4种糖苷键裂解和包括X3、X2、A3和A4在内的8种环内裂解,MS、MS2谱图分别示于图8a和8b。与dp4-1相比,都检测到了相同的糖苷键裂解(Y3、Z3、B3和C3)和环内裂解(1,5X3、2,4X2和3,5A4)。但在ESI-MS2分析过程中检测到了dp4-2独有的3种裂解方式(1,5X2、2,4A4和1,5A4),都位于含有GlcNH3+基团的葡萄糖胺上,示于图8c。
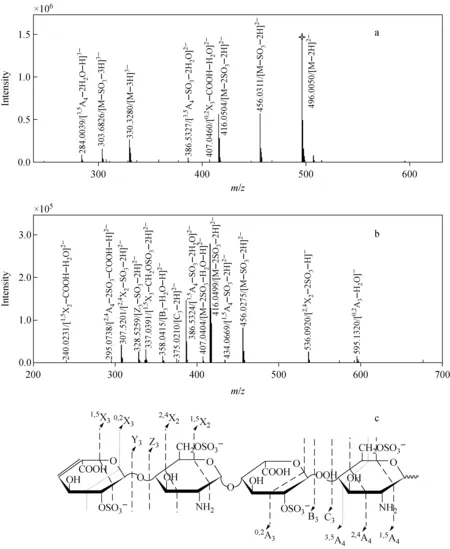
注:a.ESI-MS图谱;b.图a中m/z 496.005 0离子作为母离子的MS2图谱;
综上所述,3种dp4有相同且固定的裂解方式,但含有不同GlcNH3+残基数目的dp4能够生成不同的碎片离子,且含GlcNH3+残基数目越多,生成的碎片离子越多,这可能是GlcNH3+所带正电荷改变了环的结构,使环变得比较脆弱,在MS分析中更易裂解。
3 结论
用部分脱N位硫酸根方法制备的含不同GlcNH3+数目的肝素寡糖与生物体中存在的含GlcNH3+结构的HS具有相似的结构,这在生物体外检测功能蛋白的相互作用以及酶活实验中具有重要的价值。ESI-MS和MS2分析结果表明,含GlcNH3+数目越多,dp4能够检测到的碎片离子越多,说明该方法可用于鉴定和定量具有重要生物学功能的含GlcNH3+的HS寡糖,以及化学修饰产生的含GlcNH3+结构的寡糖,甚至它们的同分异构体。
[1] LIU D, SHRIVER Z, VENKATARAMAN G, et al. Tumor cell surface heparan sulfate as cryptic promoters or inhibitors of tumor growth and metastasis[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(2): 568-573.
[2] ZYGALAKI E, TSAROUCHA E G, KAKLAMANIS L, et al. Quantitative real-time reverse transcription PCR study of the expression of vascular endothelial growth factor (VEGF) splice variants and VEGF receptors (VEGFR-1 and VEGFR-2) in non small cell lung cancer[J]. Clinical Chemistry, 2007, 53(8): 1 433-1 439.
[3] COUCHMAN J R. Syndecans: Proteoglycan regulators of cell-surface microdomains?[J]. Nature Reviews Molecular Cell Biology, 2003, 4(12): 926-938.
[4] PERRIMON N, BERNFIELD M. Specificities of heparan sulphate proteoglycans in developmental processes[J]. Nature, 2000, 404(6 779): 725-728.
[5] CARLSSON P, PRESTO J, SPILLMANN D, et al. Heparin/heparan sulfate biosynthesis[J]. Journal of Biological Chemistry, 2008, 283(29): 20 008-20 014.
[6] KAMIMURA K, KOYAMA T, HABUCHI H, et al. Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling[J]. Journal of Cell Biology, 2006, 174(6): 773-778.
[7] JOHAN K, DOROTHE S, JINPING L, et al. Interactions between heparan sulfate and proteins: The concept of specificity[J]. The Journal of Cell Biology, 2006, 174(3): 323-327.
[8] CASU B, LINDAHL U. Structure and biological interactions of heparin and heparan sulfate[J]. Advances in Carbohydrate Chemistry and Biochemistry, 2001, 57: 159-206.
[9] ESKO J D, SELLECK S B. Order out of chaos: Assembly of ligand binding sites in heparan sulfate[J]. Annual Review of Biochemistry, 2002, 71(1): 435-471.
[10]ESKO J D, LINDAHL U. Molecular diversity of heparan sulfate[J]. The Journal of Clinical Investigation, 2001, 108(2): 169-173.
[11]LINDAHL U, KUSCHE-GULLBERG M, KJELLEN L. Regulated diversity of heparan sulfate[J]. Journal of Biological Chemistry, 1998, 273(39): 24 979-24 982.
[12]FANG C, GABRIEL S, LARS-ÅKE F, et al. Non-conserved,S-nitrosylated cysteines in glypican-1 react withN-unsubstituted glucosamines in heparan sulfate and catalyze deaminative cleavage[J]. Glycobiology, 2012, 22(11): 1 480-1 486.
[13]TOIDA T, YOSHIDA H, TOYODA H, et al. Structural differences and the presence of unsubstituted amino groups in heparan sulphates from different tissues and species[J]. Biochemical Journal, 1997, 322(2): 499-506.
[14]WESTLING C, LINDAHL U. Location ofN-unsubstituted glucosamine residues in heparan sulfate[J]. Journal of Biological Chemistry, 2002, 277(51): 49 247-49 255.
[15]REES M, PATTISON D M. Oxidation of heparan sulphate by hypochlorite: Role ofN-chloro derivatives and dichloramine-dependent fragmentation[J]. Biochemical Journal, 2005, 391(1): 125-134.
[16]WEI Z, LYON M, GALLAGHER J T. Distinct substrate specificities of bacterial heparinases againstN-unsubstituted glucosamine residues in heparan sulfate[J]. Journal of Biological Chemistry, 2005, 280(16): 15 742-15 748.
[17]SHI X, ZAIA J. Organ-specific heparan sulfate structural phenotypes[J]. Journal of Biological Chemistry, 2009, 284(18): 11 806-11 814.
[18]LIU J, SHRIVER Z, BLAIKLOCK P, et al. Heparan sulfateD-glucosaminyl 3-O-sulfotransferase-3A sulfatesN-unsubstituted glucosamine residues[J]. Journal of Biological Chemistry, 1999, 274(53): 38 155-38 162.
[19]JIAN L, ZACH S, MARSHALL P R. Characterization of a heparan sulfate octasaccharide that binds to herpes simplex virus type 1 glycoprotein D[J]. Journal of Biological Chemistry, 2002, 277(36): 33 456-33 467.
[20]SHUKLA D, LIU J, BLAIKLOCK P, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry[J]. Cell, 1999, 99(1): 13-22.
[21]VANPOUILLE C, DELIGNY A, DELEHEDDE M, et al. The heparin/heparan sulfate sequence that interacts with cyclophilin B contains a 3-O-sulfatedN-unsubstituted glucosamine residue[J]. Journal of Biological Chemistry, 2007, 282(33): 24 416-24 429.
[22]SATOMI N, EKO P, NAOKO T, et al. Heparan sulfate containing unsubstituted glucosamine residues: Biosynthesis and heparanase-inhibitory activity[J]. Journal of Biological Chemistry, 2014, 289(22): 15 231-15 243.
[23]WEI Z, DEAKIN J A, BLAUM B S, et al. Preparation of heparin/heparan sulfate oligosaccharides with internalN-unsubstituted glucosamine residues for functional studies[J]. Glycoconjugate Journal, 2011, 28(9): 525-535.
[24]WOLFF J J, CHI L, LINHARDT R J, et al. Distinguishing glucuronic from iduronic acid in glycosaminoglycan tetrasaccharides by using electron detachment dissociation[J]. Analytical Chemistry, 2007, 79(5): 2 015-2 022.
[25]HUANG R, LIU J, SHARP J S. An approach for separation and complete structural sequencing of heparin/heparan sulfate-like oligosaccharides[J]. Analytical Chemistry, 2013, 85(12): 5 787-5 795.
[26]SCHENAUER M R, MEISSEN J K, YOUJIN S, et al. Heparan sulfate separation, sequencing, and isomeric differentiation: Ion mobility spectrometry reveals specific iduronic and glucuronic acid-containing hexasaccharides[J]. Analytical Chemistry, 2009, 81(24): 10 179-10 185.
[27]SHI X, HUANG Y, MAO Y, et al. Tandem mass spectrometry of heparan sulfate negative ions: Sulfate loss patterns and chemical modification methods for improvement of product ion profiles[J]. Journal of the American Society for Mass Spectrometry, 2012, 23(9): 1 498-1 511.
[28]林江慧,张建伟,张惠芳,等. 硫酸类肝素二糖的质谱裂解规律探析[J]. 质谱学报,2015,36(2):111-119.
LIN Jianghui, ZHANG Jianwei, ZHANG Huifang, et al. Study on the fragmentation patterns of heparan sulfate disaccharides by ESI-MS[J]. Journal of Chinese Mass Spectrometry Society, 2015, 36(2): 111-119(in Chinese).
[29]林江慧,杜佳燕,付青,等. 离子肼飞行时间质谱表征硫酸软骨素硫酸角质素二糖[J]. 分析实验室,2015,34(3):270-274.
LIN Jianghui, DU Jiayan, FU Qing, et al. Characterization of disaccharides from chondroitin/dermatan sulfate by ion trap time-of-flight hybrid mass spectrometry[J]. Chinese Journal of Analysis Laboratory, 2015, 34(3): 270-274(in Chinese).
[30]HU H, HUANG Y, YU X, et al. A computational framework for heparan sulfate sequencing using high-resolution tandem mass spectra[J]. Molecular and Cellular Proteomics, 2014, 13(9): 2 490-2 502.
[31]LIANG Q T, XIAO X M, LIN J H, et al. A new sequencing approach forN-unsubstituted heparin/heparan sulfate oligosaccharides[J]. Glycobiology, 2015, 25(7): 714-725.
[32]BEAUDET J M, WEYERS A, SOLAKYILDIRIM K, et al. Impact of autoclave sterilization on the activity and structure of formulated heparin[J]. Journal of Pharmaceutical Sciences, 2011, 100(8): 3 396-3 404.
[33]FU L, LI L, CAI C, et al. Heparin stability by determining unsubstituted amino groups using hydrophilic interaction chromatography mass spectrometry[J]. Analytical Biochemistry, 2014, 461(5): 46-48.
[34]DONEANU C E, WEIBIN C, GEBLER J C. Analysis of oligosaccharides derived from heparin by ion-pair reversed-phase chromatography/mass spectrometry[J]. Analytical Chemistry, 2009, 81(9): 3 485-3 499.
[35]DOMON B, COSTELLO C E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates[J]. Glycoconjugate Journal, 1988, 5(4): 397-409.
Preparation and Characterization of Heparin Tetrasaccharide withN-unsubstituted Glucosamine Residues
LIANG Qun-tao, WEI Zheng
(InstituteofGlycobiochemistry,FuzhouUniversity,Fuzhou350002,China)

heparin; tetrasaccharide;N-unsubstituted disaccharide; liquid chromatography-ion trap/time-of-flight mass spectrometry (LC/MS-IT-TOF); fragmentation pattern
2015-12-21;
2016-03-02
国家自然科学基金(21343013)资助
梁群焘(1981—),男(汉族),福建龙岩人,博士研究生,分析化学专业。E-mail: 43415162@qq.com
魏 峥(1969—),女(汉族),福建福州人,教授,从事糖生物化学研究。E-mail: zheng0wei@hotmail.com
时间:2016-07-05;
http:∥www.cnki.net/kcms/detail/11.2979.TH.20160705.1353.022.html
O657.63
A
1004-2997(2016)06-0492-12
10.7538/zpxb.youxian.2016.0027

