基于DNA-launched的猪繁殖与呼吸综合征病毒反向遗传操作系统的建立
2016-12-09曹宗喜焦培荣谭树义叶保国亓文宝张桂红
曹宗喜,焦培荣,谭树义,叶保国,亓文宝,张桂红*
(1.海南省农业科学院畜牧兽医研究所 海南省热带动物繁育与疫病研究重点实验室,海南海口 571100;2.华南农业大学兽医学院 广东省动物源性人兽共患病预防与控制重点实验室/国家生猪种业工程中心,广东广州 510642)
研究论文
基于DNA-launched的猪繁殖与呼吸综合征病毒反向遗传操作系统的建立
曹宗喜1,2,焦培荣2,谭树义1,叶保国1,亓文宝2,张桂红2*
(1.海南省农业科学院畜牧兽医研究所 海南省热带动物繁育与疫病研究重点实验室,海南海口 571100;2.华南农业大学兽医学院 广东省动物源性人兽共患病预防与控制重点实验室/国家生猪种业工程中心,广东广州 510642)
为构建猪繁殖与呼吸综合征病毒(PRRSV)反向遗传操作系统,将PRRSV全基因组分6段克隆,构建获得6个重组质粒,先由A1和A2片段或B1和B2片段连接构成A片段或B片段,然后D、C、B和A片段依次亚克隆入pOKq载体。在基因组5′端和3′端分别加上CMV启动子序列和BGH终止信号肽,全基因组510位核苷酸突变引入遗传标记位点FseⅠ。测序正确的全基因组质粒命名为pOK-A2BCD。再将pOK-A2BCD质粒转染BHK-21细胞,拯救病毒通过PCR、酶切、测序和间接免疫荧光试验(IFA)鉴定,结果表明拯救病毒成功,为进一步研究PRRSV致病性等相关分子机制奠定了基础。
猪繁殖与呼吸综合征病毒;反向遗传操作系统;构建
猪繁殖与呼吸综合征(Porcine reproductive and respiratory syndrome,PRRS)是由猪繁殖与呼吸综合征病毒(Porcine reproductive and respiratory syndrome virus,PRRSV)感染猪所致的一种急性传染病。该病以妊娠母猪流产、死胎、弱胎、木乃伊胎等繁殖障碍,各年龄阶段猪呼吸道症状,哺乳仔猪高死亡率、免疫抑制和持续性感染等为主要特征[1-2],是目前严重危害我国乃至世界养猪业最重要传染病。而高致病性PRRSV 2006年在我国首次出现,造成严重经济损失[3-4],其致病能力显著增强现象引起高度关注。
根据转染的核酸不同,反向遗传操作平台可以分为基于RNA-launched和基于DNA-launched两种。基于RNA-launched的反向遗传操作平台是RNA病毒基因组先经过反转录成为病毒cDNA,接着克隆到合适的转录载体上,克隆到T7或SP6转录启动子下游,在体外转录合成病毒的基因组RNA,然后再将这些体外转录本转染宿主细胞,使之在宿主细胞内包装形成具有感染性的病毒粒子,最后从细胞培养物中回收病毒,得到感染性分子克隆。基于DNA-launched的反向遗传操作平台是经反转录的cDNA克隆到表达载体上获得具有感染性的cDNA,再将该质粒转染细胞,经细胞内转录生成具有感染性的病毒RNA,获得重组病毒。与基于RNA-launched的方法相比,基于DNA-launched的拯救效率高10倍~50倍,且绕过RNA体外转录这一步骤,使得拯救病毒的过程大大减低了成本和节省了时间。
目前,反向遗传技术在PRRSV的研究中已广泛应用,对病毒蛋白功能、病毒致弱作用、基因缺失标记疫苗等研究产生巨大的推动作用[5-10]。虽然本课题组已构建了基于RNA-launched的反向遗传操作平台[11],但此平台存在着操作要求严格,成本高等不利之处,因此试图建立基于DNA-launched的感染性克隆。
1 材料与方法
1.1 材料
1.1.1 载体、质粒、毒株 由pOK12 改造具有多克隆位点的低拷贝载体pOKq、PRRSV XH-GD株病毒和其分段基因组质粒pJET-A1(q)、pJET-A2(q)、pOK-B(q)、pJET-C(q)和pJET-D(q)由华南农业大学农业部新兽药研制重点实验室保存。
1.1.2 试剂 Platinum pfx DNA聚合酶和lipofectamine 2 000为Invitrogen 公司产品;CloneJET PCR Cloning Kit购自Fermentas公司;限制性内切酶NotⅠ、XhoⅠ、AflⅡ、BamHⅠ、Hind Ⅲ、KpnⅠ均为NEB公司产品;EZNA TM Gel Extraction Kit和EZNA TM Plasmid Mini Kit购自Omega公司;Endo Free plasmid Maxi kits购自QIAGEN公司;抗PRRSV N蛋白单克隆抗体(MAb)由中国农业科学院上海兽医研究所童光志研究员惠赠。
1.2 方法
1.2.1 构建策略 PRRSV全长cDNA的构建见图1,先由A1和A2片段或B1和B2片段连接构成A片段或B片段,然后D、C、B和A片段依次亚克隆入pOKq载体。根据这一构思设计引物:以pcDNA3.1-myc-His为模板,设计引物扩增CMV基因和BGH基因;以PRRSV XH-GD株的全基因组序列和本实验室保存的PRRSV XH-GD株分段基因组质粒pJET-A1(q)和pJET-D(q)的序列为模板[11],设计D片段的回复突变引物,将RNA-launched的反向遗传操作平台引入的遗传标记NotⅠ突变回来;同时设计A1片段的引物在PRRSV全基因组510位核苷酸突变引入遗传标记FseⅠ。为了便于区分拯救病毒和亲本毒,基于引入的遗传标记设计1对鉴定引物(表1),由上海英潍捷基生物技术有限公司合成。
1.2.2 融合CMV启动子的A1片段基因的扩增 以质粒pcDNA3.1-myc-His为模板,用引物CMV-F和CMV-R对CMV基因进行PCR扩增。以PRRSV分段基因组质粒pJET-A1(q)为模板,用引物A1a-F2和A1a-R或A1b-F和A1b-R分别对A1片段基因进行定点突变PCR扩增得到A1a片段或A1b片段。PCR产物用10 g/L琼脂糖凝胶(含0.5 μg/mL EB)电泳检测和回收纯化。以得到的CMV片段、A1a片段和A1b片段为模板,用引物CMV-F和A1b-R对融合CMV启动子的A1片段基因进行融合PCR扩增。PCR产物用10 g/L琼脂糖凝胶(含0.5 μg/mL EB)电泳检测和回收纯化。
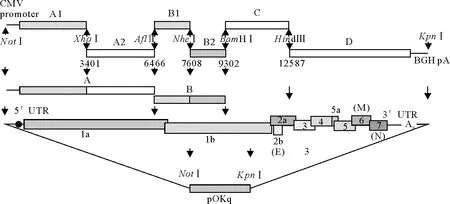
图1 PRRSV全长cDNA的构建策略

引物名称Primername引物序列(5'-3')Primersequence(5'-3')扩增片段大小/bpAmplifyinglengthCMV-FCMV-RAGTCgcggccgcCGATGTACGGGCCAGATATACTCCGAGTGGACGTGCGTCCTCCTTCGGATGCCCCTATAGTGAGTCGTATTAATTTC717A1a-F2A1a-RCGTCCACTCGGATGGCTAAGGGAGGGCCATGACGTATAGGTGTTGGCTCGCCGGCCCTGTAGATTTCAGCTGCG545A1b-FA1b-RGAAATCTACAGGGCCGGCCAACTTACAGGTGCAGGCGTGTGAGGTAACATC2961Da-FDa-RCAGGTGTTTGCCATTTTCCCAACCCCTGGCCCTAGCAGTCGGCCGCGAC1818Db-FDb-RGTCGCGGCCGACTGCTAGGGCggatgcccaggtcggaccacggggaggtggagatgccatgccgaccTTTTTTTTTTTTTTTTTTTTTTTTT1014BGH-FBGH-RacctgggcatccgaaggaggacggacgtccactcggatggctaagggagtgcGCCTCGACTGTGCCTTCTAGTAGTCggtaccCAGAAGCCATAGAGCCCAC298Det-FDet-RCTGAGCTTGGGGTGCTGATGGCGAATTTCTTTGTACTG743pJET+pJET-AACTTGGAGCAGGTTCCATTCTTAATATACAGCCTGAAAATC254空载体BlankvectorpOK+pOK-TCAGGCGTGGAATGAGACAAACAAGCTCATTCGCCATTCAGGCT485空载体Blankvector
1.2.3 融合BGH终止信号肽的D片段基因的扩增 以质粒pcDNA3.1-myc-His为模板,用引物BGH-F和BGH-R对BGH基因进行PCR扩增。以PRRSV分段基因组质粒pJET-D(q)为模板,用引物Da-F和Da-R或Db-F和Db-R分别对D片段基因进行定点突变PCR扩增得到Da片段或Db片段PCR产物用10 g/L琼脂糖凝胶(含0.5 μg/mL EB)电泳检测和回收纯化。以Da片段、Db片段和BGH片段为模板,用引物Da-F和BGH-R对融合BGH终止信号肽的D片段基因进行融合PCR扩增。PCR产物用10 g/L琼脂糖凝胶(含0.5 μg/mL EB)电泳检测和回收纯化。
1.2.4 质粒pJET-A1和pJET-D的构建 将1.2.2和1.2.3得到的PCR纯化产物分别与pJET1.2/blunt载体连接。取5 μL连接产物转化大肠埃希菌DH5α后,用灭菌牙签挑取可疑菌落于含氨苄青霉素的LB液体培养基中,于37℃振荡培养4 h~6 h,取适量菌液进行PCR鉴定。PCR鉴定的阳性重组菌液送上海英潍捷基生物技术有限公司测序进一步验证,测序正确的重组质粒命名分别为pJET-A1和pJET-D。
1.2.5 PRRSV全长cDNA的构建 先由A1和A2片段或B1和B2片段连接构成A片段或B片段,然后D、C、B和A片段依次亚克隆入pOKq载体(图1)。构建的质粒经PCR鉴定和双酶切鉴定。鉴定阳性的重组菌液送上海英潍捷基生物技术有限公司测序进一步验证,测序正确的重组质粒命名分别为pOK-A2BCD。
1.2.6 病毒拯救 将质粒pOK-A2BCD转染BHK-21细胞,采用Lipofectamine 2000脂质体转染,转染后72 h,冻融3次,4℃、7 000 r/min离心7 min,取上清,接种Marc-145细胞,连续传12代。拯救的病毒命名为rXH-GD。
1.2.7 拯救病毒的鉴定 取第3代的病毒细胞培养物悬液进行RNA的提取和DNaseⅠ消化,再取2 μg RNA进行反转录。以得到的cDNA为模板,Det-F和Det-R为引物进行PCR扩增。PCR产物用10 g/L琼脂糖凝胶(含0.5 μg/mL EB)电泳检测和回收纯化。将纯化后的PCR产物送上海英潍捷基生物技术有限公司测序鉴定,同时进行FseⅠ酶切鉴定。
将质粒pOK-A2BCD转染BHK-21细胞72 h后,分别以鼠抗PRRSV N蛋白单克隆抗体和Anti-Mouse IgG/DyLight488为一抗和二抗,参考曹宗喜等[12]的方法进行IFA检测。
1.2.8 拯救病毒的稳定性 将1.2.6得到的病毒,选择第1、3、6、9和12代病毒进行TCID50测定。
1.2.9 拯救病毒的生长曲线 测定rXH-GD 第3代病毒和亲本毒XH-GD第8代病毒的生长曲线。
2 结果
2.1 融合CMV启动子的A1片段基因的扩增
以质粒pcDNA3.1-myc-His为模板,用引物CMV-F和CMV-R对CMV基因进行PCR扩增。经10 g/L琼脂糖凝胶电泳,可见相应的目的条带,CMV基因的扩增产物长度约为720 bp,与预期大小相符(图2A)。以质粒pJET-A1(q)为模板,用引物A1a-F2和A1a-R或A1b-F和A1b-R分别对A1片段基因进行定点突变PCR扩增得到A1a片段或A1b片段。PCR产物经10 g/L琼脂糖凝胶电泳,可见相应的目的条带,分别约为0.5 kb和3.0 kb的扩增产物,与预期大小相符(图2B)。以得到的CMV片段、A1a片段和A1b片段为模板,用引物CMV-F和A1b-R对融合CMV启动子的A1片段基因进行融合PCR扩增。经10 g/L琼脂糖凝胶电泳,可见约为4.2 kb的扩增片段,与预期大小相符(图2D)。
2.2 融合BGH终止信号肽的D片段基因的扩增
以质粒pcDNA3.1-myc-His为模板,用引物BGH-F和BGH-R对BGH基因进行PCR扩增。经10 g/L琼脂糖凝胶电泳,可见相应的目的条带, BGH基因的扩增产物长度约为300 bp,与预期大小相符(图2A)。以PRRSV分段基因组质粒pJET-D(q)为模板,用引物Da-F和Da-R或Db-F和Db-R分别对D片段基因进行定点突变PCR扩增得到Da片段或Db片段。PCR产物经10 g/L琼脂糖凝胶电泳,可见相应的目的条带,分别约为1.8 kb和1.0 kb的扩增产物,与预期大小相符(图2C)。以得到的BGH片段、Da片段和Db片段为模板,用引物Da-F和BGH-R对融合BGH终止信号肽的D片段基因进行融合PCR扩增。经10 g/L琼脂糖凝胶电泳,可见约为3.1 kb的扩增片段,与预期大小相符(图2D)。
2.3 pOK-A2BCD质粒的鉴定
以CMV-F和CMV-R、BGH-F和BGH-R为引物进行PCR鉴定pOK-A2BCD质粒。经10 g/L琼脂糖凝胶电泳,可见约为720 bp或300 bp的扩增片段,与预期大小相符(图3A)。同时,进行NotⅠ和XhoⅠ双酶切鉴定,经10 g/L的琼脂糖凝胶电泳,可见约为10.6 kb的片段(载体pOKq和PRRSV B1部分片段、C片段、D片段的双酶切条带)、4.1 kb的片段(PRRSV A1片段的双酶切条带)和3.5 kb的片段(PRRSV A2片段和B1部分片段的限制性内切酶XhoⅠ双切条带),与预期大小相符(图3B)。鉴定阳性的重组菌液送上海英潍捷基生物技术有限公司进行测序进一步验证,测序正确的重组质粒命名分别为pOK-A2BCD。
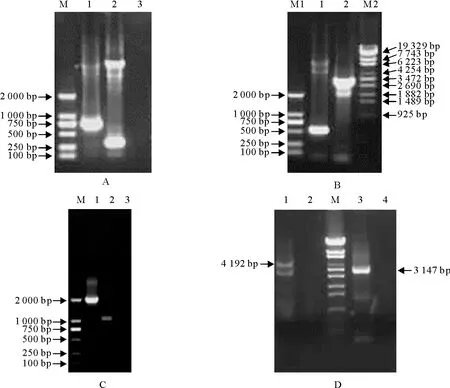
A.CMV和BGH基因PCR扩增图。M.DNA 标准DL 2 000;1.CMV扩增产物;2.BGH扩增产物;3.以水为模板的阴性对照。B.A1片段基因引入遗传标记的PCR扩增图。M1.DNA标准DL 2 000;M2.λ-EcoT14 I digest DNA Marker;1.A1a扩增产物;2.A1b扩增产物。C.D片段基因恢复突变的PCR扩增图。M.DNA标准DL 2 000;1.Da扩增产物;2.Db扩增产物;3.以水为模板的阴性对照。D.融合PCR扩增图。M.λ-EcoT14 I digest DNA 标准;1.CMV和A1融合PCR扩增产物;2.以水为模板的阴性对照;3.D和BGH融合PCR扩增产物;4.以水为模板的阴性对照
A.PCR amplification of CMV and BGH genes.M.DNA Marker DL 2 000;1.CMV PCR products;2.BGH PCR products;3.Negative control.B.PCR amplification of A1 fragment of PRRSV.M1.DNA Marker DL 2 000;M2.λ-EcoT14 I digest DNA Marker;1.Amplification of A1a;2.PCR amplification of A1b.C.PCR amplification of D fragment of PRRSV.M.DNA Marker DL 2 000;1.PCR amplification of Da;2.PCR amplification of Db;3.Negative control.D.Fusion PCR amplification.M.λ-EcoT14 I digest DNA Marker;1.Fusion PCR amplification of the CMV and A1fragment of PRRSV;2.Negative control;3.Fusion PCR amplification of the BGH and D fragments of PRRSV;4.Negative control
图2 融合CMV启动子的A1片段基因和融合BGH终止信号肽的D片段基因的扩增
Fig.2 Fusion PCR amplification of the CMV and A1 fragment of PRRSV or the BGH and D fragments of PRRSV
2.4 拯救病毒的鉴定
将pOK-A2BCD质粒转染BHK-21细胞72 h后,冻融后取上清,接种Marc-145细胞,连续传3代。取第3代的样品进行PCR扩增目的条带,经10 g/L琼脂糖凝胶回收后,进行FseⅠ酶切鉴定。拯救病毒rXH-GD和亲本毒XH-GD均可以扩增出约740 bp的目的条带;拯救病毒rXH-GD的PCR产物经FseⅠ酶切后,可见约540 bp和200 bp的目的条带,与预期结果一致(图4A)。
将纯化后的PCR产物送上海英潍捷基生物技术有限公司进行测序鉴定,结果显示拯救病毒rXH-GD的全基因组第510位核苷酸为G,该位点由A变为G,对应的阅读框从AGA变为AGG,而推导氨基酸均为精氨酸(R),为沉默突变;同时,该位点突变后引入酶切位点FseⅠ(GGCCGGCC),与预期结果一致(图4B)。将pOK-A2BCD质粒转染BHK-21细胞72 h后,IFA检测可知,拯救病毒rXH-GD有绿色荧光出现(图4C)。
A.pOK-A2BCD菌液PCR鉴定图。M.λ-EcoT14 I digest DNA标准; 1~8.以BGH-F和BGH-R为引物的pOK-A2BCD菌液PCR扩增产物; 9~16.以CMV-F和CMV-R为引物的pOK-A2BCD菌液PCR扩增产物。B.pOK-A2BCD质粒酶切鉴定图。M.λ-EcoT14 I digest DNA Marker; 1.pOK-A2BCD质粒对照; 2.pOK-A2BCD质粒NotⅠ/XhoⅠ酶切产物
A.PCR amplification of pOK-A2BCD.M.λ-EcoT14 I digest DNA Marker; 1-8.PCR amplification (primer:BGH-F and BGH-R) of pOK-A2BCD; 9-16.PCR amplification (primer:CMV-F and CMV-R) of pOK-A2BCD.B.Restricted enzymatic digestion of pOK-A2BCD.M.λ-EcoT14 I digest DNA Marker; 1.Plasmid pOK-A2BCD; 2.Enzyme (NotⅠ/XhoⅠ) digestion of pOK-A2BCD
图3 pOK-A2BCD质粒的构建
Fig.3 Construction of plasmid pOK-A2BCD
A.遗传标记位点区域的PCR扩增和酶切鉴定。M.DNA标准DL 2 000 ;1.rXH-GD PCR扩增产物;2.rXH-GD PCR扩增产物的酶切产物;3.XH-GD PCR扩增产物;4.XH-GD PCR扩增产物的酶切产物。B.PCR产物的测序分析。C.BHK-21转染pOK-A2BCD质粒72 h后,进行IFA分析(100×)
A.Identification of genetic marker by RT-PCR and restricted enzymes digestion.M.DNA Marker DL2000;1.PCR amplification of rXH-GD;2.Restricted enzymatic digestion of PCR amplification of rXH-GD;3.PCR amplification of XH-GD;4.Restricted enzymatic digestion of PCR amplification of XH-GD.B.Sequence analysis of PCR products.C .BHK-21 cells transfected with pOK-A2BCD examined by immunofluorescence assays 72 h postinoculation(100×)
图4 拯救病毒的鉴定
Fig.4 Identification of the rescued viruses
2.5 拯救病毒的稳定性
将拯救病毒rXH-GD在Marc-145细胞上连续传12代,均有细胞病变出现(图5)。第1、3、6、9和12代rXH-GD的病毒滴度分别为5.02、4.85、5.12、5.43和5.33,和亲本毒XH-GD的(XH-GD 8f的病毒滴度为4.80)相似。
图5 拯救病毒的细胞病变(200×)
2.6 拯救病毒的生长曲线
用104TCID50的拯救病毒rXH-GD或亲本毒XH-GD感染Marc-145细胞6、12、24、36、48、60、72 h后,收集上清进行TCID50测定。结果表明,rXH-GD和XH-GD的生长曲线相似,病毒达到最高滴度的时间相同(图6)。
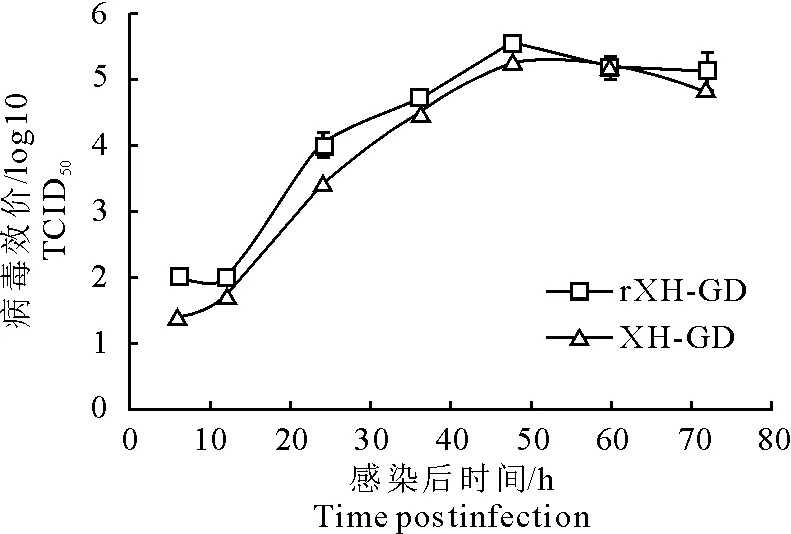
图6 拯救病毒的生长曲线
3 讨论
本研究中选择48株不同年代不同地域分离的北美型PRRSV序列和4株欧洲型PRRSV序列,通过Lasergene 7.1的MegAlign方法比对发现,PRRSV XH-GD株全基因组的第510位核苷酸在北美型PRRSV中很保守,其碱基均为A;而在此位点欧洲型PRRSV的碱基为T。同时,应用weblogo 3方法(Schneider and Stephens,1990)对这些序列进行比较分析,PRRSV基因组510位以碱基A为主(图略)。因此,选择此位点引入遗传标记位点(A510G),该位点对应的阅读框从AGA变为AGG,这2个密码子对应的氨基酸均为精氨酸(R),为沉默突变;同时,该位点突变后引入酶切位点FseⅠ(GGCCGGCC)。
本研究中所要扩增的两个融合DNA片段长度大于3 kb。对于大片段,在PCR扩增过程中,随着扩增片段长度的增加,往往会出现基因突变、缺失等保真性降低的现象。因此,在构建感染性克隆的过程中必须使用高保真DNA聚合酶来减少PCR过程中出现的基因突变。本研究选择Platinum pfx DNA聚合酶进行PCR扩增或融合PCR扩增。此外,在PCR反应过程中还应该尽可能的缩短循环数目,尽量减少核酸在紫外线下照射的时间。在后续的PCR鉴定中需要扩增更大片段,本研究选用ExTaqDNA聚合酶进行扩增,且通过两步法进行扩增。
融合PCR技术是在不需要限制性内切酶消化和连接酶处理的条件下,采用具有互补末端的引物将不同来源的扩增片段连接起来,为同源重组片段的构建提供了快速简捷的途径。本研究构建两段基因时,均采用了融合PCR技术对3个片段进行扩增,即在原有的两步反应中增加了一个步骤,将各待融合片段等摩尔混合,在不添加引物的条件下先进行11~13个循环,这种方法大大地提高了产物的特异性,减少了非特异性条带的产生[13]。
关于大片段的连接,将A片段转入质粒pOK-BCD中,即7 187 bp的片段和11 051 bp的片段相连接,本研究选择高质量的T4 DNA连接酶,以提高连接的效率;其次,减少连接体系中核酸量,增加克隆的效率;再次,调整载体DNA和插入DNA的摩尔比,本研究均取10 ng进行连接。
对于建立反向遗传操作技术平台,保持克隆序列的完整性与准确性是决定成败的关键。对于PRRSV来说,5′端序列更显重要(数据未发表)。在本研究在基因组两端均加上核酶序列,使全长质粒在细胞内转录出RNA后,在核酶的作用下直接剪切出不含外缘基因的病毒基因组。
PRRSV基因组3′端的poly A个数对病毒的拯救有重要意义。一般认为,全长cDNA克隆中的polyA的数量存在一个值,低于这个值,则其体外转录体的稳定性很差,以至无法产生有感染性的恢复病毒。目前已成功建立的PRRSV反向遗传操作技术平台中,poly A个数从21到109个不等[14-15]。本课题组建立基于RNA-launched的反向遗传操作平台时,尝试选取了39个poly A尾。因此,本研究依然选取了39个poly A尾。
郑海红等[16]采用Roche公司的FuGENE HD转染试剂转染PRRSV全长质粒到Marc-145细胞,获得拯救病毒。而本文采用Invitrogen公司的lipofectamine 2000脂质体转染PRRSV全长质粒到BHK-21细胞,获得拯救病毒。笔者曾经试图直接转染Marc-145细胞,但转染后进行IFA检测,仅可见到少量的荧光信号。与转染BHK-21细胞相比,转染效率相对较低,可能这与不同公司试剂的转染效率有关。
本研究成功拯救了PRRSV,这为进一步研究PRRSV致病性等相关分子机制奠定了基础。
[1] Renukaradhya G J,Meng X J,Calvert J G,et al.Inactivated and subunit vaccines against porcine reproductive and respiratory syndrome:Current status and future direction [J].Vaccine,2015,33(27):3065-3072.
[2] 闫晓霞,谢丽君,刘欢欢,等.PRRSV抑制Ⅰ型IFN信号通路研究进展 [J].动物医学进展,2015,36(9):93-96.
[3] Tong G Z,Zhou Y J,Hao X F,et al.Highly pathogenic porcine reproductive and respiratory syndrome,China [J].Emerg Infect Dis,2007,13(9):1434-1435.
[4] 韦 莹,洪绍锋,覃艳然,等.一株Nsp2蛋白自然缺失123个氨基酸的PRRSV分离和鉴定 [J].动物医学进展,2015,36(10):34-38.
[5] Lunney J K,Fang Y,Ladinig A,et al.Porcine reproductive and respiratory syndrome virus (PRRSV):Pathogenesis and interaction with the immune system [J].Annu Rev Anim Biosci,2016,4:129-154.
[6] Renukaradhya G J,Meng X J,Calvert J G,et al.Live porcine reproductive and respiratory syndrome virus vaccines:Current status and future direction [J].Vaccine,2015,33(33):4069-4080.
[7] Suhardiman M,Kramyu J,Narkpuk J,et al.Generation of porcine reproductive and respiratory syndrome virus byinvitroassembly of viral genomic cDNA fragments [J].Virus Res,2015,195:1-8.
[8] Khatun A,Shabir N,Seo B J,et al.The attenuation phenotype of a ribavirin-resistant porcine reproductive and respiratory syndrome virus is maintained during sequential passages in pigs [J].J Virol,2016,90(9):4454-4468.
[9] Fan B,Liu X,Bai J,et al.Influence of the amino acid residues at 70 in M protein of porcine reproductive and respiratory syndrome virus on viral neutralization susceptibility to the serum antibody [J].Virol J,2016,13:51.
[10] Li Y,Shyu D L,Shang P,et al.Mutations in a highly conserved motif of nsp1β protein attenuate the innate immune suppression function of porcine reproductive and respiratory syndrome virus [J].J Virol,2016,90(7):3584-3599.
[11] 吴欣伟,熊永忠,秦宏阳,等.猪繁殖与呼吸综合征病毒XH株感染性克隆的构建[J].中国预防兽医学报,2011,33(2):97-100.
[12] 曹宗喜,焦培荣,林哲敏,等.Marc-145细胞CD163基因克隆及其真核表达质粒构建[J].东北农业大学学报,2013,44(6):28-31.
[13] 李 敏,杨 谦.一种高效构建同源重组DNA片段的方法-融合PCR [J].中国生物工程杂志,2007,27 (8):53-58.
[14] Li Z,Wang G,Wang Y,et al.Rescue and evaluation of a recombinant PRRSV expressing porcine interleukin-4 [J].Virol J,2015,12:185.
[15] Tian D,Meng X J.Amino acid residues Ala283 and His421 in the RNA-dependent RNA polymerase of porcine reproductive and respiratory syndrome virus play important roles in viral ribavirin sensitivity and quasispecies diversity [J].J Gen Virol,2016,97(1):53-59.
[16] 郑海红,张可煜,周 艳,等.携带PCV2 ORF2的感染性PRRSV克隆在Marc-145细胞表达的研究 [J].动物医学进展,2012,33(12):1-6.
Establishment of DNA-launched Reverse Genetics System of PRRSV
CAO Zong-xi1,2,JIAO Pei-rong2,TAN Shu-yi1,YE Bao-guo1,QI Wen-bao2,ZHANG Gui-hong2
(1.HainanProvincialKeyLaboratoryofTropicalAnimalReproductionandBreedingandVeterinaryMedicine,InstituteofAnimalHusbandryandVeterinaryMedicine,HainanAcademyofAgriculturalSciences,Haikou,Hainan,571100,China;2.NationalEngineeringResearchCenterforBreedingSwineIndustry/KeyLaboratoryofZoonosisPreventionandControlofGuangdongProvince,CollegeofVeterinaryMedicine,SouthChinaAgriculturalUniversity,Guangzhou,Guangdong,510642,China)
To establish a reverse genetic system of porcine reproductive and respiratory syndrome virus (PRRSV),an DNA-launched (plasmid DNA transfection-based) reverse genetics system was developed for PRRSV by introduction of ribozyme elements at both termini of the viral genomic cDNA that were placed under the control of a CMV promoter.The strategy for construction of full-length cDNA clones:Capital letters (A1,A2,B1,B2,C,D) represent 6 overlapping fragments amplified from XH-GD genome according to the unique restriction enzyme cleavage sites in viral cDNA.An enzyme cleavage siteNotI was added to 5′end of A fragment,while 41 nucleotides of polyadenosine tail followed by BGH polyadenylation signal andKpnI were added to 3′end of D fragment by PCR mutagenesis.TheFseI in fragment A1 was created by mutation to be the genetic marker.A CMV promoter with T7 promotor preceded the viral genome.The fragments were inserted into the low-copy vector pOKq in the order of D to A.The completed full-length clone was named pOK-A2BCD.BHK cells were transfected with pOK-A2BCD,then the rescued viruses were identified by RT-PCR,restricted enzyme digestion,sequencing and IFA assays.The establishment of the reverse genetics system of PRRSV laid the foundation for further study on the molecular basis of the pathogenicity of PRRSV.
PRRSV; reverse genetics; establishment
2016-03-07
国家自然科学基金项目(31272564);现代农业产业技术体系专项资金项目(CARS-36);海南省科学事业费项目(13-214002-0002)
曹宗喜(1982-),男,河南新乡人,博士,副研究员,主要从事兽医传染病学研究。*通讯作者
S852.659.6
A
1007-5038(2016)11-0001-08
