制备方法对ZnO-ZrO2催化剂上乙醇转化制异丁烯反应的影响
2016-12-06刘田薛芳琦乐英红华伟明高滋复旦大学化学系上海市分子催化和功能材料重点实验室上海200433
刘田 薛芳琦 乐英红 华伟明高滋(复旦大学化学系,上海市分子催化和功能材料重点实验室,上海200433)
制备方法对ZnO-ZrO2催化剂上乙醇转化制异丁烯反应的影响
刘田薛芳琦乐英红华伟明*高滋
(复旦大学化学系,上海市分子催化和功能材料重点实验室,上海200433)
采用恒pH值共沉淀法和非恒pH值共沉淀法制备了ZnO-ZrO2混合氧化物催化剂,考察了制备方法对乙醇转化制异丁烯反应的影响,并用低温N2吸附、X射线衍射、扫描电子显微镜、透射电子显微镜、X射线光电子能谱、拉曼光谱、紫外-可见漫反射光谱、NH3程序升温脱附和CO2程序升温脱附对催化剂进行了表征。研究结果表明,相比于非恒pH值共沉淀法制备的ZnO-ZrO2,恒pH值共沉淀法制备的ZnO-ZrO2具有较高的比表面积,更多的酸量和碱量,从而表现出更好的乙醇转化制异丁烯催化性能。在450℃和乙醇质量空速0.2 h-1的反应条件下,两种催化剂的乙醇转化率均为100%,恒pH值共沉淀法制备的催化剂的异丁烯得率为54.9%,明显高于非恒pH值共沉淀法制备的催化剂(45.7%),并且稳定性也是前者明显高于后者。
混合氧化物;制备方法;乙醇;异丁烯;酸碱性
0 引言
随着化石能源的日益枯竭、全球气候变暖和环境日益恶化,科学家们越来越关注于寻找可再生能源来替代化石能源,以降低人们对化石能源的依赖。生物质是一种被广泛认可的可代替化石能源生产化工产品的能源,因为它是一种维持CO2平衡的中性能源载体,并且是最丰富的可再生自然资源之一,其来源非常方便[1-2]。在过去的十几年里,将生物质转化为生物燃料受到了人们的广泛关注,其中生物乙醇是生物质发酵最主要的产物[3-8]。许多科学家认为将生物乙醇转化为具有高附加值的化学产品将更具有经济效益,并且还能进一步减少CO2的排放,因此将生物乙醇转化为价值更高的化学品已经成为很多科学家的研究目标[9-13]。随着催化剂的发展,将乙醇转化为其它化工产品已有诸多文献报道[14],例如氢气[15-18]、乙烯[19-22]、丙烯[23-27]、异丁烯[28-29]、1,3-丁二烯[30-34]等等。
异丁烯是一种非常重要的有机化工原料,如:异丁烯经二聚和加氢得到的异辛烷可以作为汽油调和剂,提高汽油的辛烷值;异丁烯三聚得到的三聚异丁烯可以用作发动机燃料添加剂;异丁烯和乙醇反应得到的乙基叔丁基醚可以用作汽油添加剂;异丁烯聚合可以得到丁基橡胶。另外,异丁烯也可用来合成甲基丙烯酸酯、叔丁酚、叔丁胺、1,4-丁二醇等多种化工产品和精细化学品。随着异丁烯下游产品的开发利用,全球性异丁烯资源不足的矛盾日益突出,我国异丁烯短缺的问题也日益突出。目前,异丁烯的主要来源是石脑油蒸气裂解制乙烯装置的副产C4馏分、炼厂流化催化裂化(FCC)装置的副产C4馏分、Halcon法环氧丙烷合成中的副产物叔丁醇以及异丁烷脱氢。这些生产技术均源自不可再生的化石能源。
在乙醇转化制低碳烯烃的研究报道中,ZnOZrO2混合氧化物表现出最高的异丁烯得率[14]。有关乙醇转化制异丁烯的报道较少。Wang等[28-29]以碳为模板,合成了ZnO-ZrO2纳米混合氧化物,发现该混合氧化物对乙醇转化制异丁烯反应具有较好的催化性能,异丁烯得率可达理论得率的约80%,即异丁烯实际得率约为54%。本文用简单的共沉淀法制备了ZnO-ZrO2混合氧化物催化剂,将其用于乙醇转化制异丁烯反应,结合表征结果探讨了制备方法(即恒pH值共沉淀法和非恒pH值共沉淀法)影响催化剂性能的原因,类似的研究尚未见文献报道。
1 实验部分
1.1催化剂制备
将17.498 g ZrO(NO3)2·7H2O和1.449 g Zn(NO3)2·6H2O溶于200 mL水中,搅拌下将其与6 mol·L-1氨水同时逐滴加入100 mL pH=8的氨水中,控制整个滴加过程的pH值恒定在8,滴加结束后室温下静置陈化24 h。所得的Zn(OH)2-Zr(OH)4沉淀经过滤、洗涤、110℃干燥24 h后,于550℃静态空气中焙烧6 h,即制得ZnO-ZrO2混合氧化物,这是用恒pH值共沉淀法制备得到的混合氧化物,记为ZnOZrO2-C。非恒pH值共沉淀法制备ZnO-ZrO2混合氧化物(标记为ZnO-ZrO2-V)的差别在于,搅拌下将6 mol·L-1氨水逐滴加入ZrO(NO3)2·7H2O和Zn(NO3)2· 6H2O的混合溶液中,直至最终溶液的pH值为8,其它制备过程同上。2个样品中Zn与Zr的物质的量之比均为1∶10。
1.2催化剂表征
用X射线衍射(XRD)对样品的物相进行分析。采用Bruker D8型X射线衍射仪,Cu Kα射线(λ= 0.154 056 nm),Ni滤光片,管电压40 kV,管电流30 mA,扫描范围20°~80°,扫描速度4°·min-1。N2吸-脱附实验在Micromeritics ASAP 2010型物理吸附仪上测试。测试前样品在300℃下脱气6 h,然后在液氮温度吸附N2,用BET公式计算比表面积。催化剂的元素组成用电感耦合等离子体原子发射光谱(ICP-AES)测定,采用Perkin-Elmer Optima 8000型等离子光谱仪。扫描电子显微镜(SEM)照片在荷兰Phenom Prox扫描电子显微镜上拍摄。透射电子显微镜(TEM)照片在荷兰FEI Tecnai G2F20 S-TWIN透射电子显微镜上拍摄,拍摄前,将样品在无水乙醇中用超声波处理2 min后分散在微栅上。
样品的酸性由NH3程序升温脱附(NH3-TPD)测定。样品用量0.3 g,置于样品管中,通入流速为30 mL·min-1的He,程序升温速率为10℃·min-1,升温至500℃,保持2 h,然后降温至120℃,吸附NH3至饱和,用He吹扫2 h,以10℃·min-1的速率升温至525℃,用气相色谱仪记录NH3-TPD图。同时用液氮冷阱收集脱附的NH3,并用气相色谱定量分析。样品的碱性由CO2程序升温脱附(CO2-TPD)测定。样品用量0.3 g,置于样品管中,通入流速为30 mL·min-1的He,程序升温速率为10℃·min-1,升温至500℃,保持2 h,然后降温至50℃,吸附CO2至饱和,用He吹扫2 h,以10℃·min-1的速率升温至525℃,用气相色谱仪记录CO2-TPD图。同时用液氮冷阱收集脱附的CO2,并用气相色谱定量分析。
X射线光电子能谱(XPS)在Perkin-Elmer PHI 5000C ESAC型X射线光电子能谱仪上测定,Al Kα射线,以C1s(284.6 eV)为基准进行结合能校正。拉曼(Raman)光谱在法国HORIBA JobinYvon XploRA型激光拉曼光谱仪上测定,用功率为20 mW的氩离子激光器激发,激发波长632.8 nm,扫描范围200~1 200 cm-1。固体紫外-可见漫反射光谱用日本Shimadzu带有积分球的UV-2450型紫外-可见分光光度计测定,标准样品为BaSO4。
1.3催化剂性能评价
乙醇转化制异丁烯反应在常压固定床微型反应器中进行,催化剂用量0.8 g,反应温度450℃。乙醇水溶液(乙醇与水的物质的量之比为1∶5)由微量进样泵打入,于200℃预热气化,和N2在气化室混合均匀后进入反应器中,乙醇、水与N2的物质的量之比为1∶5∶44,乙醇的质量空速为0.2 h-1。反应前催化剂在450℃用N2活化1 h。反应产物用两台气相色谱仪在线分析,一台气相色谱仪装有FID检测器和PoraPLOT Q毛细管柱(50 m×0.32 mm×10 μm),用于检测碳氢化合物和丙酮等含氧化合物。另一台气相色谱仪装有TCD检测器和2 m长的TDX-01填充柱,用于检测CH4和CO2,检测前反应产物经-3℃的冷阱除掉大部分水。米混合氧化物时,Zn与Zr的物质的量之比低于1∶6.5的混合氧化物也没有出现ZnO晶体的XRD衍射峰。XRD结果表明2种催化剂的晶相结构相同,混合氧化物中的ZrO2均以四方晶相存在。2种催化剂的BET比表面如表1所示,恒pH值共沉淀法制得的ZnO-ZrO2-C催化剂其比表面(55 m2·g-1)明显高于非恒pH值共沉淀法制得的ZnO-ZrO2-V催化剂(26 m2·g-1)。
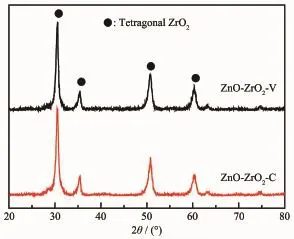
图1 ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的XRD图Fig.1XRD patterns of ZnO-ZrO2-C and ZnO-ZrO2-V catalysts
2.2SEM和TEM
图2是ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的扫描电镜照片。由图可见,2种催化剂的颗粒都没有规整的形貌,颗粒大小也差别较大,小颗粒尺寸为
2 结果与讨论
2.1催化剂的晶相结构与比表面积
图1为ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的XRD图。由图可知,2种催化剂的XRD谱一致,在衍射角(2θ)为30.2°、35.3°、50.4°和59.7°处出现了明显的衍射峰,分别对应四方相ZrO2的(111)、(200)、(220)和(131)晶面的衍射,并且2种催化剂的衍射峰强度大致相当。2种催化剂的XRD图上均未观察到ZnO晶体的衍射峰,说明ZnO在ZrO2表面高度分散[28]。Wang等[28-29]报道以碳为模板合成ZnO-ZrO2纳0.7~3.8 μm,ZnO-ZrO2-C催化剂的大颗粒尺寸为5.4~11.5 μm,ZnO-ZrO2-V催化剂的大颗粒尺寸为6.2~15.4 μm。

图2 ZnO-ZrO2-C(a)和ZnO-ZrO2-V(b)催化剂的扫描电镜照片Fig.2SEM images of ZnO-ZrO2-C(a)and ZnO-ZrO2-V (b)catalysts

表1 催化剂的比表面、酸量和碱量Table 1Surface area,acidity and basicity of the catalysts
ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的高分辨透射电镜照片如图3所示。从图中可以看出存在一些晶格条纹,其晶面间距分别为0.295和0.296 nm,与四方晶相ZrO2的(111)晶面间距十分吻合。

图3 ZnO-ZrO2-C(a)和ZnO-ZrO2-V(b)催化剂的高分辨透射电镜照片Fig.3HRTEM images of ZnO-ZrO2-C(a)and ZnO-ZrO2-V(b)catalysts
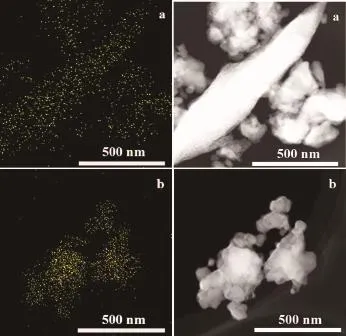
图4 ZnO-ZrO2-C(a)和ZnO-ZrO2-V(b)催化剂中的Zn元素的面扫描图(左列)和对应的HAADF STEM图(右列)Fig.4EDX elemental mapping of Zn(left column)and corresponding HAADF STEM images(right column)of ZnO-ZrO2-C(a)and ZnO-ZrO2-V(b) catalysts
用EDX测定了ZnO-ZrO2-C和ZnO-ZrO2-V催化剂中Zn元素的分布,结果见图4。从图中可以看出,总体而言,Zn在两种催化剂中均匀分布。Zn在ZnO-ZrO2-C催化剂中分散得更开,表明Zn在该催化剂中分散得更均匀。由EDX测得的ZnO-ZrO2-C和ZnO-ZrO2-V催化剂中Zn与Zr的物质的量之比分别为1∶10.1和1∶10.5(表2);由ICP测得的Zn与Zr的物质的量之比分别为1∶9.9和1∶10.3(表2),均与投料比1∶10很接近。
2.3XPS
ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的XPS结果见表2。Zn2p3/2峰的结合能分别为1 022.0 eV和1 021.8 eV,Zr3d5/2峰的结合能均为182.2 eV,表明催化剂中Zn和Zr的价态分别为+2价和+3价[35]。ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的表面Zn/Zr物质的量之比分别为1∶9.5和1∶5.4,前者与体相Zn/Zr物质的量之比(1∶10.1)较接近,后者明显高于体相Zn/Zr物质的量之比(1∶10.5),表明ZnO-ZrO2-V催化剂中的Zn在表面富集,也就是说,ZnO-ZrO2-C催化剂中的表面Zr浓度高于ZnO-ZrO2-V催化剂。
2.4Raman光谱和紫外-可见漫反射光谱

图5 ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的Raman光谱Fig.5Raman spectra of ZnO-ZrO2-C and ZnO-ZrO2-V catalysts

表2 催化剂的XPS、EDX和ICP结果Table 2XPS,EDX and ICP results of the catalysts
ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的Raman光谱如图5所示。位于267、318、465、609和646 cm-1的拉曼峰是四方相ZrO2的特征峰[36]。恒pH值共沉淀法制得的ZnO-ZrO2-C催化剂其拉曼峰强度高于非恒pH值共沉淀法制得的ZnO-ZrO2-V催化剂,这与前者较高的表面Zr浓度(见XPS结果)有关。Raman光谱没有观察到ZnO的拉曼峰,这可能与ZnO在混合氧化物中高度分散和ZnO含量较低有关。Raman光谱结果与XRD结果相一致。
图6是ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的紫外-可见漫反射光谱。2种催化剂均在209 nm有一个强的吸收峰,归属为四方相ZrO2中氧到锆的电荷转移[37]。2种催化剂的紫外-可见漫反射光谱上均未观察到单斜相ZrO2的吸收峰(约228 nm[37]),表明混合氧化物中的ZrO2均只以四方晶相存在,该结果与XRD和Raman光谱结果相一致。从图中还可以看出,2种催化剂在250~350 nm波长范围内均有吸收,这可能与ZrO2中的缺陷有关[38],另一方面,也与ZnO在该波长范围内有吸收有关[39]。
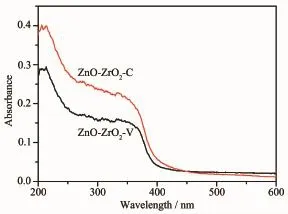
图6 ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的紫外可见漫反射光谱Fig.6Diffuse reflectance UV-Vis spectra of ZnO-ZrO2-C and ZnO-ZrO2-V catalysts
2.5NH3-TPD和CO2-TPD
图7是ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的NH3-TPD图。2种催化剂均有1个低温脱附峰和1个高温脱附峰,表明催化剂表面存在弱酸性位和强酸性位。ZnO-ZrO2-V催化剂的脱附峰温度分别为181和389℃,ZnO-ZrO2-C催化剂的脱附峰温度分别为187℃和327℃,后者的高温脱附峰温度低于前者,表明ZnO-ZrO2-C催化剂的强酸性位弱于ZnO-ZrO2-V催化剂。从脱附峰面积可以知道,ZnOZrO2-C催化剂的弱酸性位和强酸性位均明显多于ZnO-ZrO2-V催化剂。如表1所示,ZnO-ZrO2-C催化剂的酸量为0.149 mmol·g-1,远高于ZnO-ZrO2-V催化剂(0.050 mmol·g-1)。
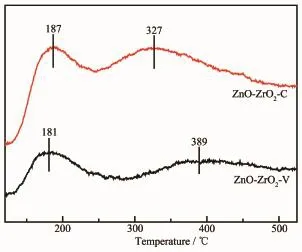
图 7ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的NH3-TPD图Fig.7NH3-TPD profiles of ZnO-ZrO2-C and ZnO-ZrO2-V catalysts
图8是ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的CO2-TPD图。ZnO-ZrO2-V催化剂只在98℃出现1个脱附峰,表明该催化剂表面只存在弱碱性位。ZnO-ZrO2-C催化剂在102℃出现1个更大的脱附峰,表明该催化剂表面的弱碱性位明显多于ZnOZrO2-V催化剂。而且ZnO-ZrO2-C催化剂还在313℃出现1个弱脱附峰,表明该催化剂表面还存在一些强碱性位,从脱附峰面积可以看出,其数量远远少于弱碱性位。如表1所示,ZnO-ZrO2-C催化剂的碱量为0.037 3 mmol·g-1,远高于ZnO-ZrO2-V催化剂(0.011 3 mmol·g-1)。

图8 ZnO-ZrO2-C和ZnO-ZrO2-V催化剂的CO2-TPD图Fig.8CO2-TPD profiles of ZnO-ZrO2-C and ZnO-ZrO2-V catalysts
2.6催化性能
于450℃考察了催化剂对乙醇转化制异丁烯的反应性能。异丁烯是主要的反应产物,此外还有二氧化碳、甲烷、乙烯、丙烯和丙酮。图9是反应4 h后的产物分布,2种催化剂的乙醇转化率均为100%。恒pH值共沉淀法制得的ZnO-ZrO2-C催化剂的异丁烯得率为54.9%(碳基实际得率),明显高于非恒pH值共沉淀法制得的ZnO-ZrO2-V催化剂(45.7%)。两种催化剂的乙烯、丙烯和二氧化碳得率基本相当。ZnO-ZrO2-C催化剂的甲烷得率(5.5%)略高于ZnO-ZrO2-V催化剂(3.2%),但前者的丙酮得率(0.7%)明显低于后者(11.5%)。
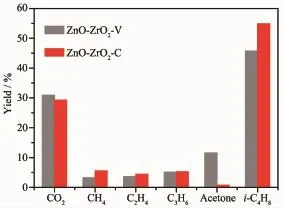
图9 ZnO-ZrO2-C和ZnO-ZrO2-V催化剂上乙醇转化制异丁烯的产物分布Fig.9Product distribution of conversion of ethanol to isobutene over ZnO-ZrO2-C and ZnO-ZrO2-V catalysts
Wang等[28-29]提出了乙醇转化制异丁烯的反应路径:

总反应:

乙醇在碱性位和酸性位的协同催化作用下脱氢生成乙醛。一般来说,碱性有利于脱氢反应,而酸性有利于脱水反应,因此,催化剂的碱性位增加,酸性位减少可抑制乙醇脱水生成乙烯,促进乙醇脱氢生成乙醛(方程式(1))。乙醛通过Aldol缩合和分解反应得到丙酮(方程式(2)),碱性有利于该反应的进行。丙酮在酸性位的催化下通过缩合、脱水和异亚丙基丙酮表面物种的分解转化成异丁烯(方程式(3))。因此,乙醇转化制异丁烯反应同时需要酸性位和碱性位。恒pH值共沉淀法制得的ZnO-ZrO2-C催化剂其酸性位和碱性位明显多于非恒pH值共沉淀法制得的ZnO-ZrO2-V催化剂,因而前者的异丁烯得率明显高于后者。ZnO-ZrO2-V催化剂的丙酮得率相对较高,这是由于该催化剂的酸性位相对较少。
进一步于450℃考察了催化反应的稳定性。异丁烯得率随反应时间的变化如图10所示。在所考察的反应时间内乙醇转化率都为100%。ZnO-ZrO2-C催化剂在24 h内异丁烯得率基本保持稳定,随后异丁烯得率缓慢下降,反应60 h后由起始的约55%降为约48%。ZnO-ZrO2-V催化剂在8 h内异丁烯得率基本保持稳定,随后异丁烯得率快速下降,反应26 h后由起始的约46%降为约5%。表明ZnOZrO2-C催化剂对乙醇转化制异丁烯的反应稳定性明显高于ZnO-ZrO2-V催化剂。异丁烯得率下降是由于催化剂积碳引起的,积碳覆盖了一部分酸性位,使得中间产物丙酮不能有效地转化为异丁烯[29]。
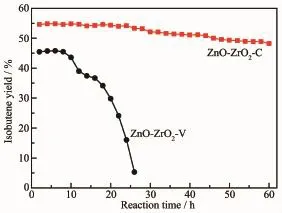
图10 ZnO-ZrO2-C和ZnO-ZrO2-V催化剂上乙醇转化制异丁烯的得率随反应时间的变化Fig.10Isobutene yield for ethanol conversion over ZnOZrO2-C and ZnO-ZrO2-V catalysts as a function of reaction time
3 结论
根据研究结果发现,ZnO-ZrO2混合氧化物中的ZrO2仅以四方晶相存在,ZnO在ZrO2表面高度分散,采用恒pH值共沉淀法制备的混合氧化物中Zn元素的分布更为均匀。XPS结果显示采用非恒pH值共沉淀法制备的混合氧化物中Zn元素在表面富集。恒pH值共沉淀法制备的催化剂比非恒pH值共沉淀法制备的催化剂具有更大的比表面、更多的表面酸性位和碱性位,从而表现出更好的乙醇转化制异丁烯催化性能。在450℃和乙醇质量空速0.2 h-1的反应条件下,2种催化剂的乙醇转化率均为100%,恒pH值共沉淀法制备的催化剂的异丁烯得率(54.9%)明显高于非恒pH值共沉淀法制备的催化剂(45.7%),并且稳定性也是前者明显高于后者。
[1]Serrano-Ruiz J C,Luque R,Sepulveda-Escribano A.Chem. Soc.Rev.,2011,40(11):5266-5281
[2]van Leeuwen B N M,van der Wulp A M,Duijnstee I.Appl. Microbiol.Biotechnol.,2012,93(4):1377-1387
[3]Huber G W,Iborra S,Corma A.Chem.Rev.,2006,106(9): 4044-4098
[4]Chheda J N,Dumesic J A.Catal.Today,2007,123(1/2/3/4): 59-70
[5]Goldemberg J.Science,2007,315(5813):808-810
[6]Sánchez Ó J,Cardona C A.Bioresource Technol.,2008,99 (13):5270-5295
[7]Hahn-Hägerdal B,Galbe M,Gorwa-Grauslund M F,et al. Trends Biotechnol.,2006,24(12):549-556
[8]Alvira P,Tomas-Pejo E,Ballesteros M,et al.Bioresource Technol.,2010,101(13):4851-4861
[9]Deluga G A,Salge J R,Schmidt L D,et al.Science,2004, 303(5660):993-997
[10]Murthy R S,Patnaik P,Sidheswaran P,et al.J.Catal.,1988, 109(2):298-302
[11]Takahara I,Saito M,Inaba M,et al.Catal.Lett.,2005,105 (3/4):249-252
[12]Kunkes E L,D.A.Simonetti D A,West R M,et al.Science, 2008,322(5900):417-421
[13]Vispute T P,Zhang H,Sanna A,et al.Science,2010,330 (6008):1222-1227
[14]Sun J M,Wang Y.ACS Catal.,2014,4(4):1078-1090
[15]Dan M,Mihet M,Tasnadi-Asztalos Z,et al.Fuel,2015,147: 260-268
[16]Kim D,Kwak B S.Appl.Surf.Sci.,2015,322:736-746
[17]Bonmati E,Casanovas A,Angurell I,et al.Top.Catal., 2015,58(2/3):77-84
[18]Li T T,Zhang J F,Xie X M,et al.Fuel,2015,143:55-62
[19]Kagyrmanova A P,Chumachenko V A,Korotkikh V N,et al. Chem.Eng.J.,2011,176:188-194
[20]Chen G W,Li S L,Jiao F J,et al.Catal.Today,2007,125 (1/2):111-119
[21]Zaki T.J.Colloid Interface Sci.,2005,284(2):606-613
[22]Bi J D,Guo X W,Liu M,et al.Catal.Today,2010,149(1/2): 143-147
[23]Inoue K,Okabe K,Inaba M,et al.React.Kinet.Catal.Lett., 2010,101(1):227-235
[24]Iwamoto M,Mizuno S,Tanaka M.Chem.Eur.J.,2013,19 (22):7214-7220
[25]Iwamoto M.Catal.Today,2015,242:243-248
[26]Hayashi F,Tanaka M,Lin D M,et al.J.Catal.,2014,316: 112-120
[27]Mizuno S,Kurosawa M,Tanaka M,et al.Chem.Lett.,2012, 41(9):892-894
[28]Sun J M,Zhu K K,Gao F,et al.J.Am.Chem.Soc.,2011, 133(29):11096-11099
[29]Liu C J,Sun J M,Smith C,et al.Appl.Catal.A,2013,467: 91-97
[30]Kitayama Y,Michishita A.J.Chem.Soc.Chem.Commun., 1981,9:401-402
[31]Gruver V,Sun A,Fripiat J J.Catal.Lett.,1995,34(3/4):359-364
[32]Makshina E V,Janssens W,Sels B F,et al.Catal.Today, 2012,198(1):338-344
[33]Ezinkwo G O,Tretjakov V F,Talyshinky R M,et al.Catal. Commun.,2014,43:207-212
[34]Lewandowski M,Babu G S,Vezzoli M,et al.Catal.Commun., 2014,49:25-28
[35]Velu S,Suzuki K,Gopinath C S.Phys.Chem.Chem.Phys., 2002,4(10):1990-1999
[36]Ji W J,Hu J Q,Chen Y.Catal.Lett.,1998,53(1/2):15-21
[37]Li M J,Feng Z H,Xiong G,et al.J.Phys.Chem.B,2001, 105(34):8107-8111
[38]Newmark A R,Stimming U.Langmuir,1987,3(6):905-910
[39]Zhang P,Shao C L,Li X H,et al.J.Hazard.Mater.,2012, 237:331-338
Influence of Preparative Method on ZnO-ZrO2Catalyst for Ethanol Conversion to Isobutene
LIU TianXUE Fang-QiYUE Ying-HongHUA Wei-Ming*GAO Zi
(Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Department of Chemistry,Fudan University,Shanghai 200433,China)
ZnO-ZrO2mixed oxide catalysts were prepared using either a constant or variable pH value coprecipitation method.The obtained catalyst samples were characterized using N2adsorption,X-ray diffraction, scanning electron microscopy,transmission electron microscopy,X-ray photoelectron spectroscopy,Raman spectroscopy,ultraviolet-visiblediffusereflectance,NH3temperature-programmeddesorptionandCO2temperature-programmed desorption.Compared to the ZnO-ZrO2mixed oxide prepared using the variable pH value method,the corresponding mixed oxide prepared using the constant pH value method had a higher surface area and a higher number of acid sites and basic sites,thus exhibiting a superior catalytic performance.When the reaction temperature was 450℃and the weight hourly space velocity of ethanol was 0.2 h-1,the conversion of ethanol for both catalysts was 100%.The ZnO-ZrO2catalyst prepared using the constant pH value method gave 54.9%yield of isobutene,which was obviously greater than the yield obtained using the corresponding catalyst prepared using the variable pH value method(45.7%).Moreover,the former catalyst exhibited obviously higher stability than the latter one for the isobutene production from ethanol conversion.
mixed oxide;preparative method;ethanol;isobutene;acidity and basicity
O614.24+1;O614.41+2
A
1001-4861(2016)08-1391-07
10.11862/CJIC.2016.177
2016-01-31。收修改稿日期:2016-05-11。
国家自然科学基金(No.21273043)和上海市科委资助项目(No.13DZ2275200)资助。
*通信联系人。E-mail:wmhua@fudan.edu.cn
