乙醇为抑制剂时磺酸树脂催化异丁烯叠合反应的理论研究
2021-02-03肖之敏洪学思周晓龙
刘 静,肖之敏,洪学思,周晓龙
(1.华东理工大学化工学院,上海 200237;2.中国石化上海石油化工研究院)
我国具有丰富的C4资源,催化裂化、蒸汽裂解、煤制烯烃等过程都会产生大量的C4烃,主要包括1-丁烯、2-丁烯、异丁烯、异丁烷、正丁烷等[1]。异丁烷和正丁烯可以通过直接烷基化反应来生产高辛烷值汽油添加组分——三甲基戊烷[2-3];异丁烯可以与甲醇通过醚化反应生产甲基叔丁基醚(MTBE)[4]。MTBE也曾作为高辛烷值添加组分加入到汽油池中,但是后来发现MTBE会污染地下水,欧洲部分国家已经禁止使用MTBE[5],导致大批MTBE生产装置闲置。因此,如何合理利用生产MTBE的原料异丁烯是目前石化行业面临的一个重要课题。探究C4烃类的转化规律,提高C4烃的高效利用具有广阔的应用前景。
异丁烯叠合反应的主要产物是二异丁烯(DIB),DIB经加氢后可得到异辛烷,其研究法辛烷值为100,且具有良好的抗爆性,是一种优良的高辛烷值清洁汽油调合组分[6-7]。而且,将MTBE的生产装置稍加改造即可用于异丁烯叠合反应,如此既可提高异丁烯的利用价值,又可降低DIB的生产成本。异丁烯叠合反应属于酸催化反应,遵守碳正离子机理[8-9]。在反应过程中,一分子异丁烯首先与催化剂的酸中心作用,形成叔碳正离子;叔碳正离子再与另一分子异丁烯叠合,形成C8碳正离子;该碳正离子既能脱去氢质子生成DIB,也能继续与异丁烯分子反应生成C12或C16碳正离子,进而生成三聚体或四聚体。基于该反应机理,在异丁烯叠合反应过程中不可避免地会产生三聚体或四聚体。三聚体加氢产物的沸点为170~180 ℃,处于汽油馏程(终馏点低于205 ℃)的末端,其加入量需要严格控制,否则会影响汽油的蒸发性能;而四聚体加氢产物的沸点为230~250 ℃,已超出了汽油的馏程范围,不能添加到汽油中[10]。因此,如果要得到高品质的汽油添加组分,必须抑制三聚体或四聚体的生成,提高二聚体的选择性。有研究表明,在异丁烯叠合反应中添加醇类(甲醇、乙醇或叔丁醇)可以显著提高二聚体的选择性[11-13]。本课题组也曾研究过以磺酸树脂为催化剂时,乙醇对混合C4叠合反应体系中异丁烯和1-丁烯转化率以及产物分布的影响,结果表明,添加乙醇可以显著提高二聚体的选择性以及二聚体中三甲基戊烯的含量,有利于提高产品质量[14],但未曾看到关于该反应体系的理论计算研究。
1 计算模拟方法
△Eads=E-EPh-SO3H-EEtOH
(1)
式中:△Eads为乙醇在磺酸基上的吸附能,kJmol;EEtOH为优化后乙醇的能量,kJmol;EPh-SO3H为优化后苯磺酸的能量,kJmol;E为优化后吸附态的能量,kJmol。
本研究所有的建模与计算均是在Gaussian09软件上完成。
2 结果与讨论
2.1 磺酸树脂模型的建立
在异丁烯叠合反应的实验研究中,本课题组曾用到Amberlyst-15和DH-2磺酸树脂[14-15]。它们均属于大孔磺酸树脂,是由磺化的苯乙烯和二乙烯基苯交联而成的一种聚合物,其中磺酸基是树脂的活性中心[16]。因此,在计算吸附能和反应活化能之前,需要先建立合理的磺酸树脂模型。如果构建相应的高聚物模型,计算结果会更加准确,但是由于该模型的原子数较多,会使得计算量非常大,耗费时间较长。Ma等[17]用密度泛函理论研究Amberlyst-15树脂催化苯酚和烯烃烷基化反应的机理时,用苯磺酸代表磺酸树脂来建模进行计算,所得到的计算结果与实验结果基本匹配,这说明在进行磺酸树脂的相关理论计算时,可以选择苯磺酸作为模型。这样不仅可以真实地反映不同物种与活性中心之间的相互作用,还可以缩短计算时间。因此,在本研究中也选用苯磺酸作为磺酸树脂的模型,优化后的苯磺酸模型如图1所示。

图1 优化后的苯磺酸模型
2.2 醚化反应活化能的计算
乙醇与异丁烯的醚化反应以乙醇分子和异丁烯分子共同吸附在磺酸基上为起点,吸附后的反应物之间进行加成反应得到吸附态的产物,最后产物从催化剂活性位点上脱附得到产物,同时催化剂的活性中心被还原。通过计算可知,乙醇与异丁烯生成乙基叔丁基醚(ETBE)的反应是协同反应。图2为醚化反应过程的吸附态反应物、过渡态及产物的结构示意;图3为醚化反应过程的能量变化,其中 ini,ts-ini,ts,pro分别表示起始反应物、中间体、过渡态及产物。表1为反应物、吸附态、过渡态及产物的几何构型参数(键长)。

图2 乙醇与异丁烯醚化反应过程涉及的结构示意
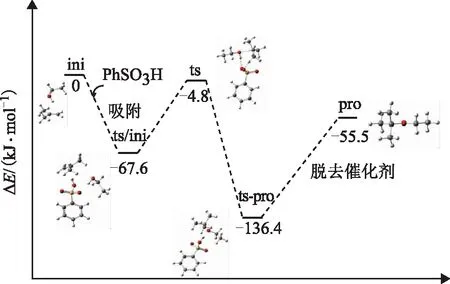
图3 醚化反应过程的能量变化
结合图2、图3及表1对醚化反应过程进行分析。首先乙醇和异丁烯共同吸附在苯磺酸上,体系的能量降低67.6 kJmol,形成稳定的吸附态,然后克服62.8 kJmol的能垒形成过渡态。在反应过程中,首先,磺酸基上的H37进攻异丁烯双键上富电子的C25,磺酸基上的O14—H37键长由0.099 961 nm不断拉长至0.146 426 nm,导致O14—H37键断裂,释放出H37;异丁烯的C25C28双键键长由0.134 526 nm伸长至0.141 016 nm,形成叔丁基碳正离子。碳正离子很容易进攻共吸附的乙醇分子上电负性较大的O24,使得O24与碳正离子C28之间的距离不断缩短,进而形成共价键O24—C28,键长为0.250 058 nm;与此同时,乙醇分子上的O24—H16键长由0.096 996 nm不断拉长至0.098 637 nm,导致键的断裂,释放出H16,该质子与磺酸基上的O15结合,形成O15—H16新键,树脂的活性中心被还原,同时形成了吸附在活性中心上的醚化产物(ETBE)。由过渡态到吸附态产物,释放了131.6 kJmol的能量,这也说明乙醇与异丁烯的醚化反应是一个放热过程。最后,吸附态的产物需要再从体系中获得80.9 kJmol的能量后从活性中心上脱附。

表1 醚化反应过程中反应物、吸附态、过渡态及产物的几何构型参数(键长) nm
2.3 异丁烯叠合反应活化能的计算
在进行异丁烯叠合反应的相关理论计算时,遵循了Eley-Rideal(E-R)反应机理。该机理认为异丁烯叠合反应的发生首先是异丁烯化学吸附在磺酸基上形成烷氧基中间体,然后再与异丁烯分子反应形成DIB,反应在一个活性中心上完成。图4和图5分别为烷氧基中间体的形成过程及异丁烯叠合反应过程中所涉及到的吸附态反应物、过渡态和产物的结构示意,图6为异丁烯叠合反应过程中的能量变化,表2为异丁烯叠合反应过程中反应物、吸附态反应物、过渡态及产物的几何构型参数。

图4 烷氧基中间体形成过程涉及的结构示意

图5 异丁烯叠合反应过程涉及的结构示意

图6 异丁烯叠合反应过程的能量变化
异丁烯分子首先被物理吸附在苯磺酸上,体系的能量降低44.8 kJmol,吸附态的异丁烯容易被磺酸基上的羟基H质子化,进而形成烷氧基中间体。该过程需要克服74.9 kJmol的能垒。根据优化后的分子模型可知,磺酸基上的28H到异丁烯两个双键碳(C16和C19)的距离分别为0.208 711 nm和0.231 927 nm,因而H28更容易进攻取代基较少的C16。在过渡态的形成过程中,磺酸基上的H28进攻异丁烯双键上C16形成叔丁基碳正离子,该碳正离子的C19很容易进攻磺酸基上电负性较大的O15。C19和O15的距离由0.237 622 nm缩短至0.148 845 nm,形成O15—C19烷氧键;同时C16—C19的键长逐渐拉长至0.152 642 nm,碳碳双键变成单键,最终形成烷氧基中间体。

表2 异丁烯叠合反应过程中反应物、吸附态反应物、过渡态及产物的几何构型参数(键长) nm
在进行异丁烯叠合反应时,游离的异丁烯分子先物理吸附在所形成的烷氧基中间体(ts1-pro)附近,体系的能量降低37.9 kJmol,得到较稳定的吸附态。烷氧基中间体与吸附的异丁烯分子发生反应,需要克服167.8 kJmol的能垒得到ts2过渡态,然后释放166.4 kJmol的能量得到吸附态的产物(ts2-pro),这也说明异丁烯叠合反应是放热反应。根据过渡态的虚频振动方向,推测在ts2过渡态的形成过程中,烷氧中间体上的O15—C19键长由0.149 441 nm不断拉长至0.377 868 nm,烷氧键断裂形成碳正离子,碳正离子C19会进攻所吸附的另一分子异丁烯双键上的C29,使得异丁烯分子的C29—C32键长拉长至0.138 842 nm;同时C32—C37单键的键长缩短变成双键,H39与磺酸基的O13成键,磺酸基活性中心被还原,并得到吸附在磺酸基上的叠合产物(DIB)。最后,从体系中吸收40.0 kJmol的能量后,产物从活性中心上脱附。
对比醚化反应和异丁烯叠合反应,醚化反应的活化能为62.8 kJmol,异丁烯叠合反应的活化能为167.8 kJmol,前者的活化能比后者低105.0 kJmol,说明醚化反应比叠合反应更容易发生。此外,叠合产物DIB从活性中心上脱附需要40.0 kJmol的能量,醚化产物ETBE的脱附需要80.9 kJmol的能量。相对于DIB,ETBE需要获得更多的能量才能从活性中心上脱附,这可能是因为ETBE中的O与磺酸基上的羟基H存在氢键相互作用,因此生成的ETBE可能并不能及时从活性中心上脱附使活性中心得到还原,而是仍占据在活性中心上,使有效的活性中心数量减少。这可能也是引入乙醇后二聚选择性有所提高的一个原因。
2.4 乙醇的作用机理分析
乙醇与异丁烯的醚化反应是可逆反应,当该反应达到平衡时,体系中仍会有未转化的乙醇存在。体系中游离的乙醇与树脂的磺酸基之间由于存在氢键相互作用会吸附在磺酸基上,而这种吸附只是一种物理吸附。根据吸附能的计算式,算出乙醇在磺酸基上的吸附能为-71.3 kJmol。由2.3节的计算结果可知,异丁烯物理吸附在苯磺酸上的吸附能为-44.8 kJmol。物理吸附态的异丁烯由于自身的电负性强容易被磺酸基上的H质子化形成烷氧基中间体,烷氧态化学吸附的吸附能为-80.2 kJmol。根据吸附能的定义可知,吸附能为负值时,其绝对值越大,说明两者的吸附作用越强,所形成的吸附态越稳定,该物质越容易吸附。虽然形成的烷氧基中间体的能量更低,但由物理吸附态转变为更稳定的化学吸附态的过程需要一定的反应时间,还要克服一定的反应能垒,因此,当乙醇存在时,优先吸附在磺酸基上的是乙醇,剩余的活性位被化学吸附态的异丁烯分子占据,乙醇的吸附能力比异丁烯强。
结合乙醇和异丁烯在磺酸基上的吸附能力以及醚化反应和叠合反应的活化能,对乙醇的作用机理进行详细的分析。在磺酸树脂催化异丁烯叠合反应体系中,当添加乙醇时,由于乙醇的吸附能力较强,故其优先吸附在活性中心上。当异丁烯分子也吸附在磺酸基附近时,由于醚化反应的活化能较低,异丁烯分子会优先与乙醇分子发生反应。醚化反应是可逆反应,当该反应达到平衡时,异丁烯分子开始发生聚合反应。由于此时树脂中的一部分活性中心及反应物中的一部分异丁烯已经参与了醚化反应,因此对于异丁烯聚合反应,有效的活性中心数量减少,反应物的实际浓度降低,使聚合反应速率降低,多聚反应受到抑制,从而提高了二聚体选择性。当醚化反应达到平衡后,体系中仍有游离的乙醇分子,由于乙醇分子与磺酸基之间存在氢键相互作用,使磺酸基为叠合反应提供氢质子的能力减弱,也就是说树脂的酸强度降低,活性降低。此外,通过计算还发现,醚化产物ETBE从活性中心上脱附所需要的能量高于叠合产物DIB的脱附,说明生成的ETBE可能并不能及时从活性中心上脱附使活性中心得到还原,而是仍占据着活性中心,这将更进一步减少有效活性中心的数量。总的来说,在异丁烯叠合反应体系中,乙醇是通过减少有效活性中心的数量、减弱树脂的酸强度以及降低反应物异丁烯的实际浓度来提高二聚体的选择性的。
3 结 论
基于密度泛函理论,利用苯磺酸来模拟磺酸树脂,推测醚化反应和异丁烯叠合反应的反应路径。醚化反应以乙醇分子和异丁烯分子共同吸附在苯磺酸上为起点,叠合反应以异丁烯形成烷氧基中间体为起点,醚化反应和叠合反应的活化能分别为62.8 kJmol和167.8 kJmol。在磺酸树脂催化异丁烯叠合反应体系中,当乙醇存在时,磺酸树脂的活性中心优先吸附乙醇,剩余的活性中心被化学吸附态的异丁烯所占据,且活化能较低的醚化反应优先发生。因此,对于异丁烯叠合反应,反应物浓度的降低、有效活性中心数量的减少以及树脂酸强度的减弱均会使聚合反应速率降低,多聚反应发生的可能性降低,从而提高二聚体的选择性。
