分子印迹固相萃取/超高效液相色谱-串联质谱法同时测定动物源性食品中11种β-受体阻断剂残留
2016-12-01邵瑞婷丁学妍
郝 杰,姜 洁,邵瑞婷,丁学妍,史 娜,路 勇
(北京市食品安全监控和风险评估中心,北京 100041)
分子印迹固相萃取/超高效液相色谱-串联质谱法同时测定动物源性食品中11种β-受体阻断剂残留
郝 杰,姜 洁*,邵瑞婷,丁学妍,史 娜,路 勇
(北京市食品安全监控和风险评估中心,北京 100041)
建立了一种分子印迹固相萃取/超高效液相色谱-串联四极杆质谱法同时测定动物源性食品中11种β-受体阻断剂残留量的方法。样品经β-葡萄糖醛苷酶/芳基硫酸酯酶酶解后,上清液调节pH值,以异丙醇-乙酸乙酯(5∶5,体积比)萃取,有机相吹干后复溶过分子印迹固相萃取柱净化。样品经BEH C18色谱柱分离,以甲醇-5 mmol/L乙酸铵水溶液(含0.1%甲酸)进行梯度洗脱,电喷雾电离正离子模式下采用多反应监测,基质加标曲线定量。11种β-受体阻断剂在0.05~10 μg/kg范围内线性关系良好,相关系数均不小于0.991,方法检出限和定量下限分别为0.02~0.1 μg/kg和0.05~0.25 μg/kg。在猪肉、猪肝基质中3个添加水平下,平均回收率分别为73.2%~108.3%及70.2%~98.2%,相对标准偏差分别为1.3%~9.5%及3.7%~9.8%。该方法快速、灵敏、准确,适用于β-受体阻断剂多组分残留的测定。
分子印迹固相萃取;液相色谱-串联质谱法;β-受体阻断剂;残留检测
β-受体阻断剂是作用于β-受体,能选择性地与β-肾上腺素受体结合,从而拮抗神经传递质和儿茶酚胺对β-受体激动作用的一类物质[1]。在临床上,针对冠心病、高血压、心律失常等心血管系统疾病的治疗发挥着极其重要的作用,是目前应用最广泛的降低心率的药物之一[2]。但β-受体阻断剂会造成对心血管系统、神经系统、消化系统等多方面的副作用,过量使用甚至会引起中毒反应[3]。
产肉动物,特别是畜类动物,自身肌体没有足够的防御机制以应对运往屠宰场途中受到的外界刺激,在运输过程途中可能会因心力衰竭而死亡[4]。这类现象会导致肌肉组织中某些代谢通路的改变,另外,在后续加工当中也存在温度或其他因素的刺激,最终会导致PSE肉(Pale soft exudative)的产生,俗称水猪肉、热霉肉。PSE肉是一种劣质猪肉,可带来巨大经济损失[5]。为了避免PSE肉的产生,在屠宰前的运输过程中,镇静类药物和β-受体阻断剂类药物会被非法注射到动物体内,以减少应激现象的发生,而这些药物往往在屠宰前数小时内被大剂量直接用药,因而相对其他兽药会造成更高的残留量和更大的风险危害[6]。另外β-受体阻断剂也是国际奥林匹克委员会禁止使用的兴奋剂[7],从食源性兴奋剂控制的角度,也需要加大动物源性食品中β-受体阻断剂的严格监控。目前国际上针对β-受体阻断剂卡拉洛尔(Carazolol)在动物组织中的残留限量已有明确要求[8]。
目前,动物源性食品中β-受体阻断剂的相关研究较少,由于其化合物结构多为碱性中等极性化合物,因此分析手段上常采用离子交换机理的固相萃取结合液相色谱-串联质谱技术[9-13]。新型的分子印迹固相萃取技术,结合了固相萃取的易操作和实用性以及免疫亲和色谱的高选择性和稳定性等特点,可实现同一类化合物高效、同步的前处理净化,特别适用于食品复杂基质中多残留组分的分析[14-15]。本文建立了分子印迹固相萃取/超高效液相色谱-串联质谱检测动物源性食品中11种β-受体阻断剂的分析方法。该方法灵敏,回收率高,稳定性好,相比于传统方法可以达到更低的检出限,且更适用于肝脏等复杂基质,可满足残留检测的需要。
1 实验部分
1.1 仪器与试剂
超高效液相色谱-串联四极杆质谱联用仪(Acuqity UPLC/Xevo TQ-S,美国Waters公司);纯水仪(Milli-Q,美国Millipore公司);高速冷冻离心机(3K18,美国Sigma公司);分子印迹固相萃取柱(Supelco Supel MIP beta-receptors 25 mg/10 mL,美国Sigma-Aldrich公司)。
拉贝洛尔、倍他洛尔、喷布洛尔、阿替洛尔、比索洛尔、阿罗洛尔、索他洛尔、纳多洛尔(CAS:42200-33-9)、奈必洛尔、卡维地洛、美托洛尔(纯度≥98%,德国Dr.Ehrenstofer GmbH);甲醇、乙腈、异丙醇、乙酸乙酯、甲酸(色谱纯,美国Thermo Fisher公司);β-葡萄糖醛苷酶/芳基硫酸酯酶(100 000 Unit/mL,德国Merck公司);乙酸铵(质谱纯,美国Sigma-Aldrich公司);乙酸钠(色谱纯,北京化学试剂厂)。
所有标准物质用甲醇溶解成1 000 μg/mL的储备液,保存于-20 ℃冰箱内,每半年对其含量进行校准。
1.2 样品制备
猪肉及猪肝样品去除脂肪进行均质,精确称取均质后的样品2.00 g于50 mL聚四氟乙烯离心管内,加入10 mL 0.2 mol/L的乙酸钠-乙酸缓冲液(pH 5.2),充分涡旋均质后,加入50 μLβ-葡萄糖醛苷酶/芳基硫酸酯酶,于37 ℃下200 r/min振荡酶解6 h以上。取出后冷却至室温,涡旋后以10 000 r/min离心10 min,取出全部上清液至另一干净离心管中,用10 mol/L氢氧化钠溶液调至pH 11.0,加入10 mL饱和氯化钠及10 mL异丙醇-乙酸乙酯(体积比5∶5)溶液,充分涡旋提取10 min,于10 000 r/min下离心,取上层有机相清液在40 ℃下氮气吹干。加入5 mL 酶解用乙酸钠-乙酸缓冲液,充分溶解残渣后上柱。
取规格为25 mg/10 mL的分子印迹柱用3 mL甲醇和3 mL水活化,溶解液全部上柱,控制流速在1滴/s,依次用3 mL水、3 mL 60%的甲醇水溶液和1 mL甲醇淋洗MIP柱,真空负压抽干后,用2 mL含1%甲酸的甲醇溶液洗脱,控制流速在1滴/s,收集洗脱液于40 ℃下用微弱氮气流吹干,用1 mL初始比例流动相复溶,过0.2 μm亲水性聚丙烯膜,供上机测定。
1.3 液相色谱-质谱条件
1.3.1 色谱条件 液相色谱柱:Waters Acquity UPLC BEH C18(100 mm×2.1 mm,1.7 μm);流动相:A为甲醇,B为5 mmol/L 乙酸铵水溶液(含0.1%甲酸);流速:0.45 mL/min;柱温:35 ℃;进样量:10 μL。梯度洗脱条件:0~0.5 min,95%~80%B;0.5~1.0 min,80%~60%B;1.0~2.5 min,60%~40%B;2.5~3.0 min,40%B;3.0~3.5 min,40%~1%B;3.5~4.5 min,1%B;4.5~5.0 min,1%~95%B;5.0~6.0 min,95%B。
1.3.2 质谱条件 电喷雾离子源,正离子模式,毛细管电压2.8 kV,离子源温度150 ℃,脱溶剂气温度500 ℃,脱溶剂气流量800 L/h,锥孔气流量150 L/h,雾化气压力7.0 bar,碰撞气流量0.17 mL/min。11种化合物的质谱参数见表1。
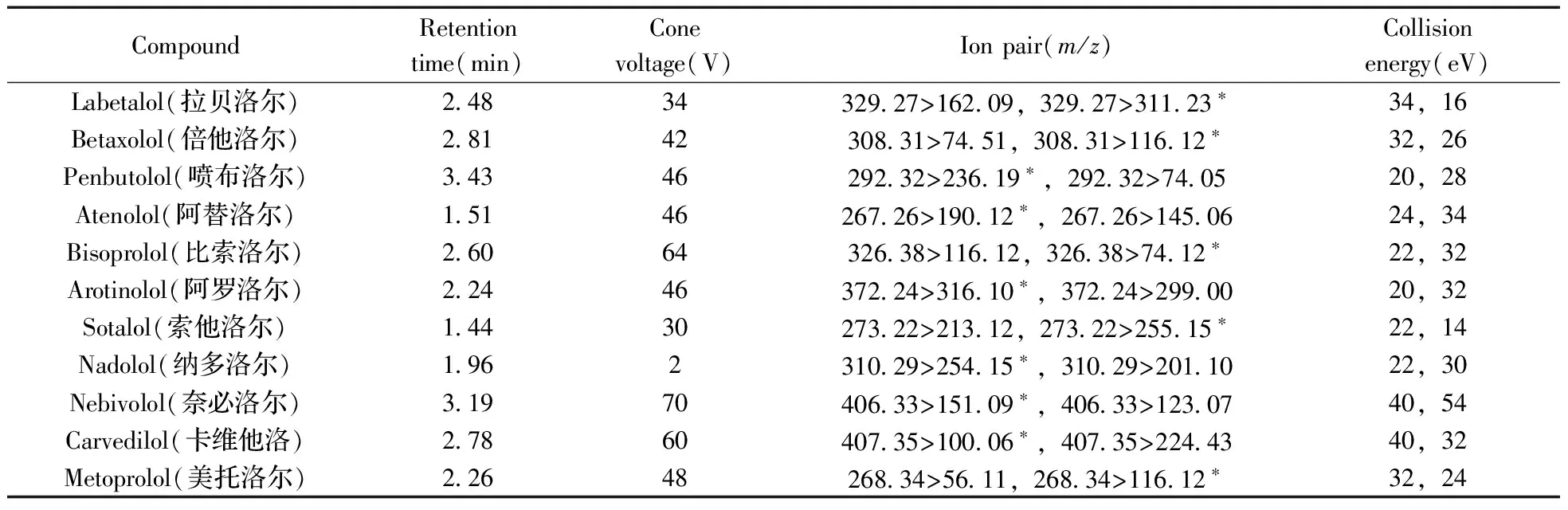
表1 11种β-受体阻断剂的色谱-质谱参数
*quantitative ion
2 结果与讨论
2.1 色谱-质谱条件的优化
选用BEH C18超高效液相色谱柱作为分离介质,对比了甲醇、乙腈与水、0.1%甲酸水溶液、5 mmol/L乙酸铵水溶液及5 mmol/L乙酸铵水溶液(含0.1%甲酸)作为流动相时的分离情况。结果表明,由于11种β-受体阻断剂均使用正离子模式检测,在流动相中加入酸可有效增强信号响应,同时加入缓冲盐可以改善峰形。因此,最终选择甲醇-5 mmol/L乙酸铵水溶液(含0.1%甲酸)作为流动相。
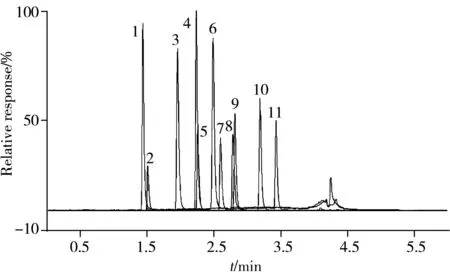
图1 11种β-受体阻断剂的色谱分离图(0.5 ng/mL)Fig.1 Chromatogram separation of 11 β-blockers at concentration of 0.5 ng/mL1.sotalol;2.atenolol;3.nadolol;4.arotinolol;5.metoprolol;6.labetalol;7.bisoprolol;8.carvedilol;9.betaxolol;10.nebivolol;11.penbutolol
使用电喷雾质谱在正离子扫描模式下,用质谱直接进样的方式,分别确定11种β-受体阻断剂的质谱参数。通过优化毛细管电压、锥孔电压和碰撞能量等参数,选取响应丰度较高的几个碎片离子作为子离子。再通过配制空白基质标准溶液,排除易受基质干扰的碎片,得到2对监测离子对,选择响应丰度较高、干扰较少的作为定量离子对。图1为优化条件下11种β-受体阻断剂的总离子流图。
2.2 样品前处理过程的优化
β-受体阻断剂的残留监测通常为痕量测定,前处理方法需达到浓缩、净化的效果。吴永宁等[16]对酶解条件进行了深入研究,具有较高的参考意义。本文主要对酶解后的提取过程与净化条件进行优化。
2.2.1 提取溶剂的选择 在酶解之后,β-受体阻断剂在酸性条件下以离子态游离于缓冲液中,将提取液调节至pH值呈碱性,使目标化合物的电离平衡向分子方向移动,目标化合物以不解离的分子状态被不互溶的有机溶剂提取,水相中留下pKa值不相同的其他可溶杂质。从提取净化的策略可知,用于提取的有机试剂的组成和配比是这一步骤中影响回收率的关键,因此对提取溶剂种类(乙酸乙酯、异丙醇、乙腈)进行了优化,并考察了其不同比例的提取效率。结果表明,使用乙腈导致两相分配不佳。而乙酸乙酯和异丙醇对11种化合物的分配各异,采取不同配比时所得到的提取效率也不同。进一步考察了不同异丙醇-乙酸乙酯比例对11种β-受体阻断剂的萃取情况,在兼顾所有化合物的原则下,最终使用异丙醇-乙酸乙酯(5∶5)进行提取。
2.2.2 净化条件的选择 对比了极性分配固相萃取柱(Waters Sep-pak C18,500 mg/6 mL)、亲水亲脂分配固相萃取柱(Waters Oasis HLB,150 mg/6 mL)、混合强阳离子固相萃取柱(Waters Oasis MCX,150 mg/6 mL)、混合强阳离子固相萃取柱(DikmaProElut PXC,150 mg/6 mL)、分子印迹固相萃取柱(SupelcoSupel MIP beta-receptors,25 mg/10 mL) 5种固相萃取柱的净化效率。结果表明,在C18柱及HLB柱上,由于是共价键结合分配机理,故回收率不理想。MCX和PXC均基于苯磺酸型强阳离子交换机理,已有报道也多使用该类型的固相萃取柱,且大部分化合物能够达到净化效果,但个别化合物(如索他洛尔)回收率不佳,仅有45.6%。MIP柱具备特异性的β-受体结构,能够特异性地结合β-受体阻断剂中的苯乙醇胺母核,使得MIP柱净化具有高度专一性和较好的回收率。对比MCX柱及PXC柱,多数化合物在MIP柱上能够达到更好的回收率,11种化合物的回收率均大于75%,同时在前处理过程中使用低温和pH值调节等步骤也能有效除去脂肪和蛋白质的干扰。因此实验选择分子印迹固相萃取柱进行净化。
2.3 方法验证
2.3.1 标准曲线与检出限 在空白样品中分别添加0,0.01,0.02,0.05,0.1,0.25,0.5,1,2,5,10 μg/kg的β-受体阻断剂混合标准物质,进行前处理和测定。以11种β-受体阻断剂的响应峰面积(Y)为纵坐标,对应浓度(X,μg/kg)为横坐标绘制工作曲线。以3倍信噪比计算方法检出限(LOD),10倍信噪比确定方法定量下限(LOQ)。11种β-受体阻断剂的线性方程、相关系数、线性范围及检出限、定量下限结果见表2。各化合物在0.05~10 μg/L范围内呈良好的线性关系,其线性相关系数(r2)为0.991 2~0.999 1。各化合物的检出限和定量下限分别为0.02~0.1 μg/kg和0.05~0.25 μg/kg。
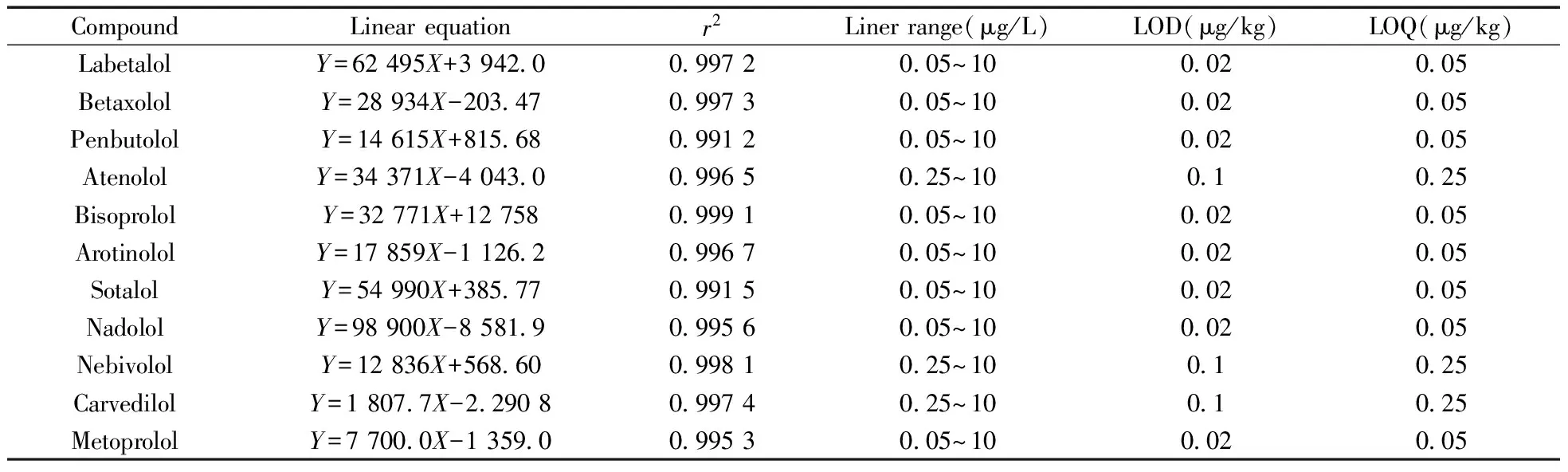
表2 11种β-受体阻断剂的线性方程、相关系数、线性范围、检出限及定量下限
2.3.2 方法的回收率与精密度 分别取动物肌肉、肝脏空白样品,添加低、中、高3个不同浓度水平的11种β-受体阻断剂混合标准溶液,优化条件下测定目标化合物。每个浓度水平平行测定6次,结果见表3。11种化合物在猪肉中的平均回收率为73.2%~108.3%,相对标准偏差(RSD)为1.3%~9.5%;在猪肝中的平均回收率为70.6%~98.2%,RSD为3.7%~9.8%。其准确度与精密度能满足残留检测的要求。
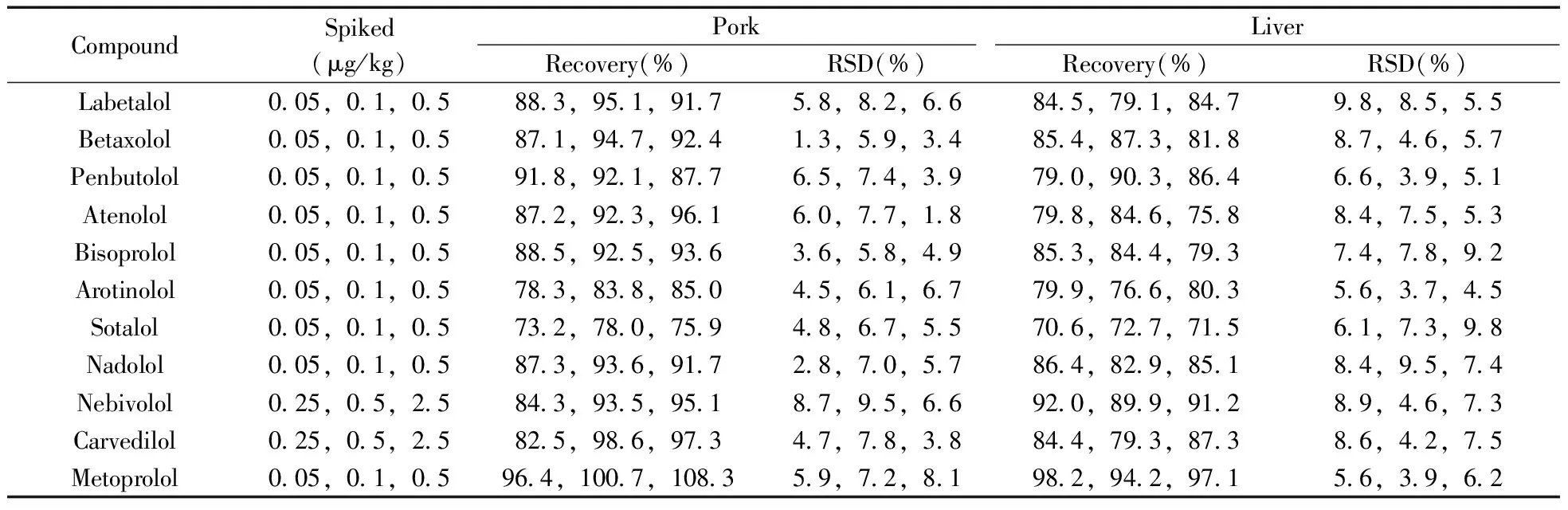
表3 11种β-受体阻断剂的回收率及相对标准偏差

图2 阳性质控样品的总离子流图Fig.2 Total ion chromatogram of positive QC sample
2.4 实际样品的测定
随机选取市售猪肉、猪肝共10份,同时做阳性质控样品。应用建立的方法对样品进行11种β-受体阻断剂的测定。结果在阳性质控样品中检出卡维他洛0.497 μg/kg(图2),其余样品均未检出。
3 结 论
本研究采用分子印迹固相萃取/超高效液相色谱-串联四极杆质谱技术,建立了猪肉及猪肝中11种β-受体阻断剂残留的同时检测方法。经方法验证,该方法检出限低、回收率高、精密度好,可用于动物源性食品中β-受体阻断剂的残留检测。
[1] Xu H,Zhang H W,Zhang G,Wang F M.J.Chin.Mass.Spectrom.Soc.(许辉,张鸿伟,张罡,王凤美.质谱学报),2015,2:1-8.
[2] Lü L.J.Pract.Med.Tech.(吕露.实用医技杂志),2006,13(9):1603-1604.
[3] Wang L J.Chin.Med.Mod.Dis.Educ.Chin.(王丽娟.中国中医药现代远程教育),2010,8(9):229.
[4] Pang G F.ModernTechniquesforResidueAnalysisofPesticideandVeterinaryDrugs.Beijing:Science Press(庞国芳.农药兽药残留现代分析技术.北京:科学出版社),1999.
[5] Quan B Z,Huang R Y,Hua X B,Chen J L,Xie D P,Zhang S M,Shao Y.JiangxiJ.Anim.Husb.Veter.Med.(全炳昭,黄仁友,花象柏,陈鹭江,谢德麃,章寿民,邵莹.江西畜牧兽医杂志),1996,1:16-19.[6] Miao H,Zou J H,Fan S,Gan L W,Zhao Y F,Wu Y N.Chin.J.Chormatogr.(苗虹,邹建宏,范赛,甘乐文,赵云峰,吴永宁.色谱),2010,28(6):572-578.
[7] World Anti-Doping Agency.The World Anti-Doping Code:The 2015 Prohibited List.[2016-03-18].https://www.wada-ama.org/en/resources/science-medicine/prohibited-list.
[8] CAC/MRL 02-2005.Maximum Residue Limits for Veterinary Drugs in Foods.Codex Alimentarius Commission.
[9] Delahaut Ph,Levaux C,Eloy P,Dubois M.Anal.Chim.Acta,2003,483(1/2):335-340.
[10] Kamila M,Andrzej P,Jan Z.Anal.Chim.Acta,2009,637(1/2):185-192.
[11] Liu J J,Zhang J,Shao B,Li Q S.Res.Environ.Sci.(柳晋杰,张晶,邵兵,李青山.环境科学研究),2009,22(7):862-867.
[12] Zhang J,Shao B,Yin J,Wu Y N,Duan H J.J.Chromatogr.B,2009,877(20/21):1915-1922.
[13] Fan S,Miao H,Zhao Y F,Chen H J,Wu Y N.J.Agric.FoodChem.,2012,60(8):1898-1905.
[14] David M G,Francisco J L,Nikola J,Laura G G,Ana M G.Anal.Chim.Acta,2015,891:321-328.
[15] Meritxell G,Tania M P,Mira P,Maria J L A,Damia B.J.Chromatogr.A,2008,1189(1/2):374-384.
[16] Wu Y N,Miao H,Fan S,Zhao Y F.Sci.Sin.:Chim.(吴永宁,苗虹,范赛,赵云峰.中国科学:化学),2009,39(8):774-784.
Determination of Eleven β-Blocker Residues in Animal Derived Foods by Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry with Molecularly Imprinted Solid Phase Extraction
HAO Jie,JIANG Jie*,SHAO Rui-ting,DING Xue-yan,SHI Na,LU Yong
(Beijing Municipal Center for Food Safety Monitoring,Beijing 100041,China)
A molecularly imprinted solid phase extraction coupled with ultra high performance liquid chromatography-tandem mass spectrometric (UPLC-MS/MS) method was developed for the determination of eleven beta blocker residues in animal derived foods.The sample was enzymed withβ-glucuronidase/aryl sulfatase.The supernatant fraction was adjusted to the optimal pH value,and then extracted with isopropanol-ethyl acetate (5∶5,by volume).The organic solvent was first evaporated to dryness, and then redissolved,finally purified by molecularly imprinted solid phase extraction.The analyte was separated with a BEH C18column by gradient elution using methanol-5 mmol/L ammonium acetate (containing 0.1% formic acid) as mobile phase,and detected by MS under ESI+ ionization and multiple reactions monitoring mode.The quantification of the analytes was performed by the matrix standard addition method.The limits of detection and limits of quantitation were in the ranges of 0.02-0.1 μg/kg and 0.05-0.25 μg/kg,respectively.The calibration curves of 11β-blockers were linear in the range of 0.05- 10 μg/kg,with correlation coefficients not less than 0.991.Under three spiked levels,the average recoveries of the analytes in pork and liver were 73.2%-108.3% and 70.2%-98.2%,respectively,with relative standard deviations of 1.3%-9.5% and 3.7%-9.8%,respectively.The method is rapid,sensitive and precise,and is suitable for the determination ofβ-blocker multi residues in animal derived foods.
molecularly imprinted solid phase extraction;liquid chromatography-tandem mass spectrometry (LC-MS/MS);β-blocker;residue determination
2016-03-24;
2016-04-18
北京市科技计划(Z161100000616007)
10.3969/j.issn.1004-4957.2016.10.010
O657.63;TQ460.72
A
1004-4957(2016)10-1278-05
*通讯作者:姜 洁,博士,高级工程师,研究方向:食品安全,Tel:010-50959673,E-mail:jybjj2004@126.com
