胰岛素基因突变: 从遗传学和β细胞生物学到临床疾病
2016-11-24MingLiuJinhongSunJingqiuCuiWeiChenHuanGuoFabrizioBarbettiPeterArvan崔景秋刘铭校译
Ming Liu, Jinhong Sun, Jingqiu Cui, Wei Chen, Huan Guo, Fabrizio Barbetti, Peter Arvan, 崔景秋,刘铭校译
·热点·译文·
胰岛素基因突变: 从遗传学和β细胞生物学到临床疾病
Ming Liu, Jinhong Sun, Jingqiu Cui, Wei Chen, Huan Guo, Fabrizio Barbetti, Peter Arvan, 崔景秋,刘铭校译
近年来,越来越多新的胰岛素基因突变开始吸引人们的注意,已证实突变位于胰岛素基因的非翻译区和前胰岛素原的编码区,包括信号肽、胰岛素链、C肽和A链,以及信号肽酶和激素原转换酶的蛋白水解剪切位点。这些突变影响胰岛细胞中胰岛素合成的不同步骤,引起胰岛素原错误折叠和早发的常染色体显性糖尿病,前胰岛素原基因突变所致的前胰岛素原加工缺陷、胰岛素原错误折叠和内质网应激可能在糖尿病发生、发展中发挥重要作用。
糖尿病;胰岛β细胞;胰岛素生物合成;胰岛素基因突变;内质网应激;错误折叠的胰岛素原
1 概述
胰岛素的作用是调节机体代谢,维持血糖平衡,在胰岛细胞中,需要30~150 min合成成熟且有生物活性的胰岛素。
(1)胰岛素前体——前胰岛素原在细胞质中翻译后,定向转位跨过内质网膜,在内质网膜的腔内侧被信号肽酶水解,形成胰岛素原。(2)在氧化的内质网环境中,胰岛素原发生氧化折叠,形成3个进化保守的二硫键(B7-A7,B19-A20和A6-A11),使得胰岛素原可以转运出内质网。(3)胰岛素原通过高尔基体的胞内运输,被激素原转换酶(PC)1/3和PC2和羧肽酶E(CPE)加工处理成C肽和成熟胰岛素,储存在胰岛素分泌颗粒中,遇刺激后释放(图1)。在1967年胰岛素原被发现之后,众多研究聚焦于胰岛素原的细胞内运输、加工处理、储存颗粒的形成和随后的分泌。
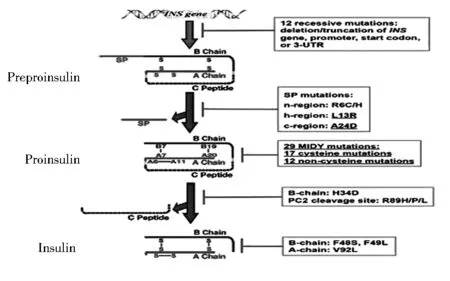
图1 胰岛素基因突变对胰岛素生物合成主要步骤的影响
胰岛素病的概念最初是用来描述罕见的与胰岛素基因突变相关的单基因成人发病糖尿病。那些突变位于胰岛素或胰岛素原的二元剪切位点,经典胰岛素基因突变损害了胰岛素与受体的结合,导致胰岛素原运输到储存颗粒的改变或者胰岛素内源性蛋白分解、加工处理的缺陷。
2007年,Stoy首先报道了10个新的导致新生儿糖尿病的胰岛素基因突变。截至目前,共有51个胰岛素基因突变可引起单基因糖尿病(表1),这些突变位于胰岛素基因的非翻译区、前胰岛素原信号肽编码区、信号肽酶的蛋白水解剪切位点、胰岛素B链、C肽、A链和PC1/3和PC2的剪切位点。临床表现谱广泛,从严重的新生儿发病到温和的成年人发病,表明不同的胰岛素基因突变体等位基因表型不同,并通过不同的机制引发糖尿病。越来越多的证据表明致糖尿病胰岛素基因突变的临床严重性与突变特性以及它们所影响的胰岛素生物合成途径的步骤相关(图1,表1)。
2 致糖尿病的胰岛素基因突变
基于遗传学结果,胰岛素基因突变可分为两大组:隐性和显性。隐性胰岛素基因突变是功能缺失的突变,包括胰岛素基因缺失或截断等,伴随胰岛素基因转录失败、mRNA不稳定或者由于缺乏天然起始密码子造成的翻译起始缺陷,从而影响胰岛素的生物合成。这些突变可能导致突变体等位基因的胰岛素产物减少。而其显性遗传模式表明剩余正常的胰岛素等位基因足以维持正常的血糖。啮齿类有两个有功能的胰岛素基因:Ins1和Ins2,研究显示Ins1或Ins2的纯合子缺失加上第二个胰岛素基因的杂合子缺失并不能导致糖尿病,再次证明即使仅存一个有功能的胰岛素等位基因,也足使血糖维持在正常范围内(虽然有功能的胰岛素等位基因缺失个体在成人时期发生糖尿病的风险可能大大增加)。大约80%(39/51)的胰岛素基因突变以常染色体显性方式遗传,其中大多数(30/39)引起青少年发病型的胰岛素基因突变糖尿病(MIDY)。对Akita鼠的研究显示,胰岛素原Ins2等位基因发生C96Y突变,导致内质网应激(ERS)和β细胞死亡。除C96Y突变外,其他导致MIDY的基因突变引起胰岛素生成缺陷的原因可能是由于胰岛细胞中共表达的突变和野生型胰岛素原分子之间异常的相互作用,突变基因阻止了共表达的野生型胰岛素原的转运,并最终导致胰岛素产量减少,表明在MIDY中,突变胰岛素原对野生型胰岛素原的显性负效应是胰岛素缺乏的关键点。

表1 胰岛素基因突变及其对胰岛素生物合成的影响
除了导致MIDY的突变外,少数(9/39)常染色体显性胰岛素基因突变与迟发型糖尿病有关,包括先前证实的经典的胰岛素病和新近发现的两个前胰岛素原信号肽突变。不像导致MIDY的基因突变,与迟发型糖尿病有关的突变似乎与内质网中的胰岛素原折叠无关,笔者描述了这一独特的胰岛素基因突变群体,大多基于已经实验证实的胰岛素生物合成过程中的细胞生物学缺陷,未经实验证实的突变则整合入与突变相关联的预测缺陷群组(图1,表1)。
3 来自胰岛素基因突变的启示:生物学缺陷和糖尿病临床表型的联系
致糖尿病的胰岛素基因突变表现出广泛的临床谱,从新生儿发生的严重胰岛素缺乏型糖尿病到迟发的轻型糖尿病,一个携带C43G突变的家族病例显示,此突变破坏了关键的链间二硫键,导致内质网中胰岛素原错误折叠,携带突变的先证者43周龄时发生了很严重的糖尿病,然而,其父亲虽然携带同样突变,却在30岁时才确诊轻微的2型糖尿病。因此,临床表现的异质性依赖于胰岛素基因突变本身的生物学行为和其他遗传环境因素。
3.1 影响基因转录和翻译的胰岛素基因突变 在胰岛细胞中,胰岛素的生物合成在转录和翻译水平上得到调节。经葡萄糖刺激后,总蛋白合成增加2~6倍,而前胰岛素原的生物合成在1 h内增加30倍,表明可能存在特殊的调控机制。几个新发现的影响转录和翻译的胰岛素基因突变从遗传学上证实了胰岛素基因非翻译区调控元件的病理生理学重要性。共12个突变以隐性方式进行遗传,其中5个位于启动子区,导致肌腱膜纤维肉瘤癌基因同系物A和神经源性分化因子-1(NEUROD1)调节的启动子区缺失或者额外的DNA结合蛋白结合位点的断裂,导致启动子活性降低90%。表明胰岛素顺式调节元件是调节胰岛素生物合成所必需的条件。位于胰岛素mRNA 3′非翻译区(3′-UTR)的c.*59 A>G突变,可以导致胰岛素mRNA严重的不稳定性,在调节胰岛素生物合成过程中起重要作用。位于前胰岛素原起始密码子第3个核苷酸的两个突变(c.3G>T和c.3G>A)破坏了前胰岛素原本身的翻译起始位点,虽然前胰岛素原第5个残基处还有另一个编码甲硫氨酸的ATG,可作为备选的起始密码子,但ATG上游的核苷酸序列并不支持翻译的起始,导致了胰岛素产量减少约80%。
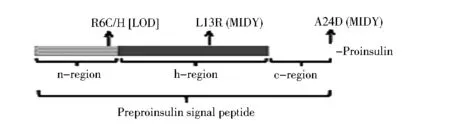
图2 与糖尿病相关的前胰岛素原信号肽突变及其3个功能区
3.2 影响前胰岛素原内质网定位和转位的胰岛素基因突变 胰岛素生物合成的第一步包括新合成的前胰岛素原从细胞质转位入内质网,此过程由前胰岛素原N端信号肽引导。前胰岛素原的信号肽有24个氨基酸,包括3个区:正电荷的n区,中央疏水的h区和一个极性的含有信号肽酶剪切位点的c区。目前,已经报道4个位于信号肽区的新突变可以引发糖尿病,分别位于3个区(图2),它们的临床表型差异很大,包括L13R/A24D引起的新生儿发病的严重胰岛素缺乏糖尿病和R6C或R6H相关的成人发病的轻型糖尿病,这些患者糖尿病的发生、发展可能存在不同的细胞学缺陷和分子机制。笔者近期发表的信号肽剪切位点A24D突变不仅导致无效的信号肽剪切,而且由于剪切发生在其他候选位点产生了异常的胰岛素原,因此虽然A24D突变前胰岛素原的胰岛素原部分具有与野生型相同的序列,未剪切和不适当剪切的前胰岛素原均可导致内质网错误折叠。凸显了信号肽剪切与下游胰岛素原折叠协调性的重要意义。
在哺乳动物细胞中,大多数新合成的分泌蛋白以共翻译的方式转位进入内质网,在此过程中,当新生多肽链的信号肽进入核糖体后,首先被信号肽识别颗粒(SRP)识别并结合,形成信号肽-核糖体-新生多肽链复合体,接着复合体通过SRP锚定到内质网膜上的SRP受体而定位于内质网膜上,随后信号肽与转位子Sec61相互作用,剩余多肽链的转位借此定向。至少有3个因素决定了信号肽序列在Sec61内的定向:h区的长度、h区侧面信号序列的电荷梯度和跨越内质网膜的转位子的电荷梯度。因为Sec61具有相对于内质网腔保守的正电荷梯度,信号肽的n区正电荷易于形成一个环,使得信号肽的N端面向胞质,C端面向内质网腔,即所谓的“正电荷在里面”法则。然而,虽然此法则是公认的前蛋白通过内质网膜转位过程中信号肽的定位原则,但分泌膜蛋白的信号肽中n区正电荷的病理生理学意义仅仅在近来才得到验证。
前胰岛素原R6C或R6H突变影响分泌蛋白的定位、转位,但其定位到胰岛素颗粒的过程并未发现缺陷,笔者研究小组也分别检测了带有大GFP(248个氨基酸)标签、小Myc标签(10个氨基酸)或者无标签的R6C或R6H突变的前胰岛素原的转位效率,结果证实带有GFP标签的R6C/H突变胰岛素原在胰岛素定位、转位过程中没有缺陷,而至少50%新合成的Myc标签/无标签的R6C/H突变胰岛素原在定位到内质网膜后不能有效地转位,提示与小分泌蛋白相比,大的分泌蛋白对信号肽n区正电荷缺失敏感性小。这个长度依赖的潜在机制,以及是否该行为是前胰岛素原所特有的,尚不明确。最近一项研究显示,多肽链的长度决定分泌蛋白通过共翻译方式还是翻译后方式进行转位,全长合成完成之前,小分泌蛋白的信号肽可能不易被SRP以共翻译的方式识别和定位。因此,定位和转位的后翻译模式可能提供一个重要的备份机制,以增强包括前胰岛素原在内的小分泌蛋白的转位效率。
在一个宫内发育迟缓女婴体内发现L13R突变前胰岛素原,其出生后次日就发生了严重的糖尿病。此突变位于前胰岛素原信号肽的h区,带电荷的精氨酸取代了疏水的亮氨酸,从而打破了h区的疏水核,可能影响分泌蛋白的SRP识别、内质网靶向定位和转位。因此,L13R突变影响新合成的前胰岛素原进入内质网的最早期步骤,然而它导致β细胞功能衰竭和糖尿病的细胞内缺陷仍然需要进一步实验验证。
3.3 影响内质网中胰岛素原折叠的胰岛素基因突变 在所有常染色体显性胰岛素基因突变中,超过70%(30/39)可影响内质网中胰岛素原正常的折叠途径(表1)。目前证实,大约半数突变可以导致内质网中胰岛素原的错误折叠,其中研究最多的是C96Y突变,由于其最初在Akita鼠中发现,故又称Akita突变。研究显示,两个Ins2等位基因中的一个携带C96Y突变,使小鼠断乳后不久即发生糖尿病,胰岛素原6个保守的半胱氨酸残基中的一个发生突变,可阻断二硫键B7-A7的形成,产生一个未配对的B7位半胱氨酸,体内、外研究均证明C96Y突变引起内质网中胰岛素原的错误折叠,诱发ERS,最终导致β细胞死亡。另一个携带C95S突变的Munich糖尿病鼠是由于N-乙基N-亚硝基脲化学诱变产生的。C95S和C96Y突变都已经在人类中被发现并可引起MIDY,所以这两种鼠都可用于研究错误折叠的胰岛素原在体内引发细胞缺陷和应激的研究。此外,还有16个突变也可产生新的未配对的半胱氨酸残基,这些突变位于本身的半胱氨酸位点,或者是突变产生了新的半胱氨酸残基,因为半胱氨酸残基中天然二硫键的形成是内质网胰岛素原折叠途径的关键步骤,所有具有未配对半胱氨酸残基的突变都可能严重影响二硫键的形成,导致胰岛素原的错误折叠。
除了产生未配对半胱氨酸外,还有12个突变可导致内质网胰岛素原的错误折叠,其中11个位于B链,1个位于前胰岛素原信号肽剪切位点,需要进一步研究点突变如何影响胰岛素原在内质网中的折叠。值得关注的是,几乎所有突变都位于前胰岛素原分子高度保守的残基,一些对胰岛素自身折叠至关重要,参与B链和A链的排列,以利于天然胰岛素原二硫键的配对。因此,即使这些突变并不直接改变参与胰岛素原正常二硫键配对的半胱氨酸残基,但A24D,H29D,L30P,G32R,L35P,R46Q和G47V突变后新合成的胰岛素原,都存在二硫键形成缺陷,导致未配对的二硫化物同分异构体增加。
3.4 影响胰岛素原加工处理和运输的胰岛素基因突变 胰岛素原经过内质网内的氧化折叠和二聚化离开内质网至高尔基体。当跨高尔基体网络腔内Zn2+浓度开始升高时,胰岛素原发生Zn2+依赖的六聚化,进入不成熟的分泌颗粒,经过PC1/3和PC2以及CPE的内源性蛋白水解剪切作用,胰岛素原六聚体被加工为成熟的胰岛素和C肽,5个胰岛素基因突变位于胰岛素原B链和C肽的连接处,或者C肽和A链之间,分别与PC1/3优先和PC2优先的剪切位点相对应。这些剪切位点突变的临床表型依据不同的残基取代而变异很大。特别是C肽和A链连接处的精氨酸被亮氨酸、组氨酸或脯氨酸所取代,可导致胰岛素原加工的缺陷,与相对无症状的高胰岛素原血症、交界性葡萄糖耐量或者迟发的轻微糖尿病相关。相反,如果在C肽-A链或C肽-B链连接处用半胱氨酸取代精氨酸,则可导致早发的严重胰岛素缺陷型糖尿病,亮氨酸R89L突变可以使胰岛素原有效地从细胞中分泌出去,而半胱氨酸R89C突变则引起胰岛素原的错误折叠并储留在内质网,因此,虽然R89C突变发生在PC2剪切位点,其致糖尿病的潜在机制并不是由于PC2介导的胰岛素原加工处理缺陷,主要是由于胰岛素原的错误折叠和内质网应激。这更彰显了内质网中胰岛素原错误折叠引发的严重病理结果。
而另一个引起高胰岛素原血症的突变源于34位天门冬氨酸取代了保守的组氨酸而发生的H34D突变,对应于B链第10个残基。对表达这一突变的转基因鼠的研究显示,超过50%的H34D突变胰岛素原可以正常转运到分泌颗粒并加工成胰岛素。然而,约15%新合成的胰岛素原从细胞中分泌,20%在β细胞降解。对短暂表达H34D突变胰岛素原的At-T20细胞的研究也获得类似的结果,在β细胞中几乎所有转运出内质网的新合成胰岛素原最终都可进入分泌颗粒,H34D突变的胰岛素原排序缺陷表明胰岛素原的结构信息在β细胞内排序、运输和储存过程中起重要作用。
3.5 影响胰岛素与受体结合的胰岛素基因突变 20世纪80年代早期就已经证实F48S、F49L和V92L[即F(B24)S、F(B25)L和V(A3)L]突变均可导致经典胰岛素病,这些突变位于胰岛素与其受体结合重要的区域。上述突变胰岛素与其受体的结合效率从野生型的0.2%到14%不等。这些突变胰岛素与其受体结合受损使胰岛素清除率显著受损,进而导致其半衰期增加,使循环中胰岛素/C肽的比例升高。这些发现证实,受体介导的胰岛素生理降解是胰岛素体内清除的主要途径。
除了受体结合缺陷,近来研究发现,与V92L或引起MIDY的突变相比,F48S突变呈现出中间表型,包括内质网氧化折叠的中等缺陷、分泌有限的减少、未折叠蛋白反应(UPR)的适度激活以及对共表达的野生型胰岛素原的部分显性负效应。研究表明,胰岛素B链第24位残基苯丙氨酸在稳定天然的疏水侧链方面起重要作用,增加了B19-A20二硫键配对的效率。来自核磁共振分光镜的计算机模型数据显示,F48S突变导致B20-B30残基结构的变异,与B19-A20二硫键配对干扰相符。相反,另一个经典的V92L突变胰岛素原的结构与野生型胰岛素相同,却仅引起轻微的热稳定性的降低。
4 胰岛素基因突变引起β细胞功能衰竭和糖尿病的潜在分子机制
4.1 内质网中错误折叠胰岛素原的细胞反应 一旦转运到内质网腔,前胰岛素原就立即经过快速的氧化折叠,加工成胰岛素原。导致常染色体显性MIDY的胰岛素基因突变,可以引起胰岛素原在内质网中的错误折叠。错误折叠的分子与内质网分子伴侣相结合,可以被内质网质控系统所识别,免疫球蛋白重链结合蛋白(BiP)是与错误折叠的胰岛素原和折叠中间体结合的主要分子伴侣之一。作为热休克蛋白70(HSP70)超家族的一员,BiP是识别未折叠和错误折叠蛋白的主要分子伴侣,通过间接促进3个UPR信号通路包括蛋白激酶样内质网激酶(PERK)、肌醇需求酶(IRE1)和活化转录因子6(ATF6)的激活,感受内质网环境变化,维持内质网稳态平衡。在正常的β细胞,3个跨膜感受蛋白的腔内区均具有与BiP结合的能力,然而,在表达致MIDY的基因突变的β细胞,BiP优先与错误折叠的胰岛素原结合,协同IRE1、PERK和ATF6的激活,启动UPR。近来研究表明,未折叠蛋白也能直接结合激活IRE1。UPR的急性活化是一种可能减轻ERS的防御机制,可以通过抑制蛋白质翻译、选择性上调内质网分子伴侣以及加速错误折叠蛋白质的逆转位,使蛋白体降解,这一过程称作内质网相关降解(ERAD)。然而,在Akita鼠(也可能在MIDY患者)中,错误折叠的胰岛素原在内质网中的持续表达导致内质网扩大,UPR活化延长,打破内质网蛋白质的动态平衡,导致慢性ERS和凋亡信号的激活,促进β细胞死亡。ERS相关的凋亡可以被C/EBP同源蛋白(CHOP:基因名称GADD153)的转录激活所介导。GADD153的靶向断裂降低了β细胞的死亡率,使杂合Akita鼠糖尿病的发生延缓8~10周。
4.2 错误折叠的胰岛素原对共表达的野生型胰岛素原的旁观效应 Akita鼠的糖尿病仅是由于ERS介导的β细胞死亡所引起的,然而如果通过阻断GADD153的转录,减少Akita鼠的β细胞凋亡,仅可以延缓但并非阻止糖尿病的发生,表明可能还有除了β细胞数量减少之外的其他因素参与了错误折叠胰岛素原导致糖尿病的发生过程。对β细胞共表达Akita突变和融合了GFP的胰岛素原的杂合鼠进行研究,发现新生Akita鼠在出现高血糖时,其β细胞数量并没有减少,Gupta等发现Akita鼠发生糖尿病时,胰岛出现了β细胞增生。最近建立了一个Akita突变的转基因猪模型,其出生后24 h内就表现出明显的高血糖-胰岛素缺乏的直接征象,然而与生后1周多的同窝对照仔相比,转基因猪的β细胞数量并没有实质差别。这些研究表明,在错误折叠的突变胰岛素原导致糖尿病的过程中,胰岛素缺乏先于β细胞数量减少。
敲除Ins1/Ins2的小鼠再与其他INS基因杂合,并不能引起糖尿病,如同人胰岛素基因突变一样,敲除其中一个胰岛素等位基因,表现隐性。因此,β细胞数量减少或者一个正常的胰岛素等位基因缺失可能都不是MIDY引发糖尿病的充分理由。如上所述,Akita鼠的β细胞并没有因表达错误折叠的突变胰岛素原而发生ERS。然而,与野生型相比,Akita鼠β细胞中所有的蛋白质包括特异的胰岛素原合成均有所减少,表明胰岛素产量的减少并非由于胰岛素原合成减少所致。脉冲-追踪实验显示,合成2 h后,大多数野生型胰岛素原加工为成熟胰岛素,而剩余部分却没被加工,发生降解,导致胰岛素产量减少。总之,对Akita鼠的研究表明,胰岛素缺乏是由于胰岛素原到胰岛素的生物合成途径缺陷所引发的。
基于上述理由,MIDY突变引起的正常胰岛素原加工过程的细胞缺陷发生在β细胞数量显著减少之前,可能是它们致胰岛素产量减少的原因。Akita鼠的大量野生型胰岛素原错误折叠,可被内质网分子伴侣BiP识别,表明错误折叠的突变胰岛素原能影响共表达的野生型胰岛素原的正常折叠,原因可能是:(1)错误折叠的突变胰岛素原引起的ERS能改变内质网环境,改变分子伴侣的利用率和氧化还原环境,甚至可能影响前胰岛素原转位的动力学以及信号肽在内质网膜的剪切,从而直接/间接影响野生型胰岛素原在内质网的正常折叠通路。然而,在用低剂量衣霉素预处理16 h诱导ERS的细胞中,野生型胰岛素原持续分泌出内质网,因此,轻微的急性ERS似乎不足以阻断胰岛素原的运输(而长时间严重的ERS对胰岛素原折叠和运输的影响仍然有待明确)。(2)错误折叠的突变胰岛素原直接影响共表达的野生型胰岛素原的内质网折叠信号通路。似乎包括了突变胰岛素原半胱氨酸残基对野生型分子间的巯基攻击,形成分子间二硫键。因为胰岛素原的全部6个半胱氨酸残基通常都会形成分子内二硫键配对,因此任何分子间二硫键都是非天然的,并会给野生型带来一个未配对的半胱氨酸残基,进一步产生不适当的相互作用,因此影响野生型胰岛素原的折叠,导致突变和野生型分子之间形成二硫键。这种异常蛋白复合体导致野生型胰岛素原储留在内质网中不能进一步转运。因此Akita突变的β细胞也能持续合成正常量的胰岛素原,只是野生型胰岛素原的内质网输出障碍直接导致了胰岛素产量的减少,这是胰岛素缺乏型糖尿病的始动因素。
错误折叠的突变胰岛素原对共表达的野生型胰岛素原的显性负效应似乎呈剂量依赖性。雄性Akita Ins2杂合鼠断乳后很快出现高血糖,而雄性Akita Ins2纯合鼠出生后即发生更严重的糖尿病。相反,当人为控制Akita突变低水平表达时(内源性Ins2不多于4%),糖尿病发生率很低(雄性低于10%),但糖耐量实验呈现糖尿病前期表现。同样,Akita突变胰岛素原水平低于内源性胰岛素基因15%的转基因猪,也没有糖尿病表型。反之,当Akita突变胰岛素原的表达继续升高达到75%时,转基因猪在生后24 h内即出现高血糖。细胞实验中也呈现剂量依赖关系,随着突变与野生型胰岛素原表达的比例逐渐下调,野生型胰岛素原的显性负效应也逐渐减少。综上,细胞和动物实验均表明致MIDY的基因突变以剂量依赖的方式起作用。
4.3 内质网中突变胰岛素原和共表达的野生型胰岛素原的相互作用 最近研究不仅证实致MIDY的基因突变对野生型胰岛素原呈剂量依赖的显性负效应,而且证实野生型对突变胰岛素原的双向效应。同一突变转染β细胞与非β细胞得到不同的结果。在转染的β细胞中,由于存在大量内源性野生型胰岛素原,研究者发现某些致MIDY基因(如R89C)的突变胰岛素原可转运出内质网,运输到胰岛素分泌颗粒。相反,在缺乏野生型胰岛素原的非β细胞,同一突变则无法转运出内质网。
更重要的是,这种营救效应似乎是蛋白特异性的,虽然野生型胰岛素原营救了某些致MIDY的突变胰岛素原,但却不能营救共表达的突变甲状腺球蛋白。相反,野生型甲状腺球蛋白也不能营救共表达的致MIDY基因突变的胰岛素原的分泌,而却能促进两个突变cog-Tg和rdw-Tg甲状腺球蛋白的分泌。这种双向效应在MIDY发生、发展中的病理生理意义仍然需要进一步评估但可能在某些遗传疾病的发病机制中起重要作用。在常染色体显性疾病如MIDY的杂合子中,占主导方向的是突变阻断了共表达的野生型蛋白,有些导致MIDY的基因突变根本不能被野生型胰岛素原营救,所以在这些杂合子患者中,没有双向效应。反之,在常染色体隐性疾病中,主导方向是野生型蛋白不仅有效地从内质网输出,它们也可以促进其二聚体突变搭档运出内质网,减轻这些错误折叠突变蛋白引起的潜在ERS。
4.4 未转位/错误转位的前胰岛素原的细胞质反应 前述3个前胰岛素原信号肽突变R6C、R6H和L13R会影响SRP的识别、内质网的靶向定位和(或)前胰岛素原通过内质网膜的转位。近来研究显示靶向定位到内质网后,R6C/H突变前胰岛素原不能有效地转位通过内质网膜,在细胞内降解。研究表明,大多数新合成的未转位R6C/H突变前胰岛素原分子被降解,然而,也有一定比例的未转位前胰岛素原在细胞内重新定位,聚集在细胞质部分的核连接部位。
错误折叠和聚积倾向的蛋白在细胞质内表达,就会发生核连接孔的聚集,细胞质蛋白稳态的紊乱和核连接处的聚集可以诱导细胞质应激,与内质网应激截然相反,R6C突变前胰岛素原导致诱导细胞质应激的主要伴侣HSP70的表达,结合错误折叠的蛋白质,防止其聚集并促进降解,更重要的是,错误折叠的蛋白质在细胞质核连接处聚集,与亨廷顿病、帕金森病、阿尔茨海默病、朊病毒病等不同神经变性疾病的发生、发展有关。R6C突变前胰岛素原导致β细胞死亡增加,促进前胰岛素原无效转位引发迟发型糖尿病。因此,不像其他常染色体显性胰岛素基因突变引起的分泌通路的缺陷,R6C/H引起的前胰岛素原细胞质的聚集似乎可以通过一个全新的机制导致β细胞功能衰竭和糖尿病。
L13R突变前胰岛素原引起的细胞缺陷还没有被实验证实,与引起迟发型糖尿病的R6C/H突变不同,L13R突变引起宫内发育迟缓,并在出生后第2天即引发严重的胰岛素缺乏型糖尿病。虽然推测L13R突变也可以影响前胰岛素原的转位,但还有其他机制参与β细胞功能衰竭和糖尿病发病。与R6C突变类似,阻断依赖SRP蛋白的转位,可诱导热休克反应,抑制细胞生长,促进细胞死亡。既然L13R突变可以影响依赖SRP的转位,L13R突变是否可导致β细胞生长停止。研究证实,同样阻断信号肽疏水核心的突变——前甲状旁腺激素原preProPTH-C18R可以导致甲状旁腺功能减退,由于突变的信号肽被SRP识别不佳而导致转位缺陷,大部分定位于内质网,诱导ERS,促进细胞死亡,以此类推,前胰岛素原L13R突变可能也通过ERS引起β细胞功能衰竭和糖尿病,但还需要进一步研究。
5 防止胰岛素生物合成早期步骤缺陷引起糖尿病的方法和策略
一半以上胰岛素基因突变(30/51)可以引起内质网中胰岛素原的错误折叠和MIDY。在过去10余年里,有关错误折叠的胰岛素原导致β细胞功能衰竭的分子机制研究取得了很大的进展,为研发防止或延缓糖尿病发病的策略奠定了基础。如上所述,虽然ERS和β细胞死亡是MIDY的标志,但更早期的事件(例如在β细胞数量大量减少之前)包括错误折叠的突变胰岛素原对野生型胰岛素原的阻断所导致的胰岛素产量的减少,可能更易于诱发胰岛素的缺乏。假定仅存一个正常的胰岛素等位基因就足以产生足够的胰岛素维持血糖正常,那么营救野生型胰岛素原不被阻断,就可能恢复足够的胰岛素产量,以延缓/防止胰岛素缺乏型糖尿病的发生。
两个特殊的方法可用于营救野生型胰岛素原不被突变胰岛素原所阻断,首先突变胰岛素原的显性负效应是剂量依赖的,如果能靶向降解突变胰岛素原,就可以使野生型胰岛素原转运出内质网。虽然泛素蛋白酶系统和自噬在Akita鼠的胰岛素原降解中起重要作用,但调节错误折叠胰岛素原降解的关键细胞内分子仍然未知。识别这些分子可能为调节错误折叠突变胰岛素原降解提供潜在的靶点,以营救野生型胰岛素原。
其次,设法改善野生型胰岛素原的在内质网中的氧化折叠,因为突变和野生型胰岛素原间异常的分子间二硫键利于突变胰岛素原的显性负效应,加速野生型胰岛素原氧化折叠的方法可以促进其折叠,限制其与共表达的突变胰岛素原相互作用,允许其在因突变储留之前逃离内质网,蛋白质二硫化异构酶(PDI)和内质网oxidoreductin-1(ERO1)在维持氧化环境促进二硫键配对中起重要作用。近来研究显示,ERO1的过度表达能改善野生型胰岛素原的氧化折叠,即使它与导致MIDY的基因突变共表达,也可使野生型胰岛素原从内质网中输出显著增加,并伴随着胰岛素产量的增加。
ERO1引起的内质网过度氧化可以导致ERS。然而研究显示,在表达突变胰岛素原的细胞中,尽管过度表达野生型或激活ERO1可增加内质网腔的氧化,但并不增加突变胰岛素原诱导的ERS,甚至似乎还导致突变胰岛素原诱导的ERS的显著减少。原因可能源自胰岛素原氧化折叠的改善和从内质网中输出量的增加,但是增加内质网腔氧化对β细胞功能的长期效应仍然需要进一步体内实验评估。
另外,PDI的过度表达并不能改善胰岛素原的氧化折叠或者增加胰岛素的产量。因为PDI不能调节胰岛素原的营救,所以在胰岛素原二硫键的形成过程中,PDI可能并不起作用。
6 结论
新的胰岛素基因突变的发现重新激发了人们对胰岛素在β细胞生物合成过程的兴趣,这些突变为探寻糖尿病发生、发展中分子和细胞缺陷提供了独特的工具。虽然还有许多问题悬而未决,但已经开始深入理解胰岛素生物合成早期事件缺陷导致的β细胞衰竭和糖尿病的分子机制,包括前胰岛素原的定位、通过内质网膜的转位、胰岛素原信号肽的剪切和新生胰岛素原分子在内质网中的折叠(图3),仍然需要进一步研究胰岛素原在β细胞内质网中的折叠途径,因为研究表明错误折叠的胰岛素原可能把它的错误折叠传播给旁观的野生型胰岛素原分子。包括错误折叠和旁观的野生型胰岛素原的化学计量法、内质网腔的氧化还原环境以及β细胞中ERAD结构的活性等都是治疗MIDY的新方法。与此同时,胰岛素原的错误折叠可能发生在没有任何突变的病理条件下,MIDY可能是2型糖尿病发生ERS的极端案例,可以为研究2型糖尿病ERS提供有力的工具,所有这些联系有待进一步探索。
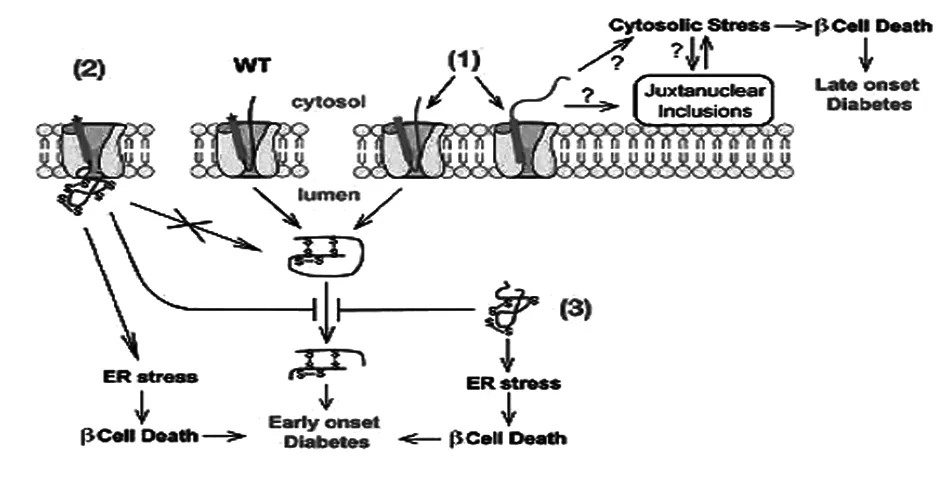
图3 胰岛素生物合成早期事件缺陷引发糖尿病和β细胞功能衰歇的模式图
(本文译自:INS-gene mutations:From genetics and beta cell biology to clinical disease. Mol Aspects Med, 2015,42:3-18.DOI:10.1016/j.mam.2014.12.001. 已取得期刊及原作者的授权)
国家自然科学基金资助项目(81370895,81570699);天津市科委应用基础与前沿技术研究重点项目(11JCZDJC185000)
10.3760/cma.j.issn.1673-4157.2016.02.013
300052 天津医科大学总医院内分泌代谢科
刘铭,Email:ming165@hotmail.com
2015-09-28)
