同晶置换改性FAU分子筛结构及吸附性能的密度泛函理论研究
2016-11-23杨丙星叶丽萍顾慧劼徐华胜2李会英4
杨丙星,叶丽萍,顾慧劼,徐华胜2,3,罗 勇,李会英4
(1.聚烯烃催化技术与高性能材料国家重点实验室,上海200062;2.上海化工研究院,上海200062;3.上海绿强新材料有限公司,上海201608;4.上海应用技术学院应用催化研究所,上海201418)
同晶置换改性FAU分子筛结构及吸附性能的密度泛函理论研究
杨丙星1,2,叶丽萍1,2,顾慧劼1,2,徐华胜2,3,罗 勇1,2,李会英4
(1.聚烯烃催化技术与高性能材料国家重点实验室,上海200062;2.上海化工研究院,上海200062;3.上海绿强新材料有限公司,上海201608;4.上海应用技术学院应用催化研究所,上海201418)
采用密度泛函理论(DFT)方法,考察了八面沸石(FAU)型分子筛β笼孔道结构内含氧化合物(甲醇、二甲醚、丙醛)的吸附,并进一步计算研究了Zn,Ca同晶置换改性的作用机理.研究结果表明,β笼孔道结构内,Al原子为甲醇、二甲醚和丙醛的吸附活性位,Si原子无吸附活性.Zn,Ca掺杂的β笼结构内,正2价的Zn和Ca掺杂替换正3价的Al,导致邻近的Si原子位置形成缺电子空穴,增强了甲醇、二甲醚和丙醛的吸附,而杂原子Zn和Ca本身并没有吸附活性.
密度泛函理论;FAU分子筛;掺杂;吸附
煤基甲醇制烯烃工艺(MTO)由于原材料和生产工艺的不同,烯烃产物中含有少量的含氧化合物,而少量含氧化合物的存在会破坏催化剂的活性中心,影响产品质量.X型分子筛具有天然矿物八面沸石(FAU)的骨架结构,具有较大的孔体积(约占50%)、比表面积和三维微孔结构,将X型分子筛应用于MTO工艺中脱除醇醚研究具有现实意义.
夏思奇等[1]通过引入异丙醇控制合成出具有较大孔容和较小晶粒尺寸的13X分子筛,并将其应用于甲醇、二甲醚和丙醛等含氧化合物的脱除,吸附性能良好.研究发现,离子交换改性[2~5]、脱铝改性[6~8]、杂原子同晶置换改性[9~12]及孔道修饰改性[13,14]等方法可以改变分子筛的结构从而影响其性能,如Zhang等[5]利用密度泛函理论方法计算研究了碱金属阳离子对Cu+Y分子筛活性中心周围电子环境和催化甲醇制碳酸二甲酯性能的影响,结果表明,离子交换改性方法可有效提高分子筛催化剂的反应活性.碱改性处理的HZSM⁃5催化剂,介孔数量增加,平均孔径增大,总酸量降低,B酸/L酸降低,优化了催化剂的芳构化协同催化作用,提高了低碳烃芳构化活性、选择性和稳定性[15].
杂原子同晶置换改性可在分子筛原有骨架中引入特定的非金属或金属原子,从而改变分子筛的氧化还原性能及催化活性等,避免了负载可能存在的活性物种流失,非骨架物种堵塞微孔孔道等问题[16].同晶置换改性方法广泛应用于ZSM⁃5[9,17,18],ZSM⁃22[19],SBA⁃15[11]等不同种类的分子筛,常用改性金属主要包括Zn[18],Ga[16,17],La[20],Ca[18,21],Fe[18,21,22]及Cr[22,23]等.如Yang等[24]制备了Zn掺杂的HZSM⁃5分子筛,并用NaOH后处理得到介孔Zn/HZSM⁃5分子筛,有效改善了HZSM⁃5分子筛的抗烧结性能,甲醇制芳烃的收率和寿命都明显提高.Zhou等[25]利用一锅法制备出碱金属元素(Mg,Ca,Sr,Ba)掺杂改性的全硅分子筛(AeS⁃1),发现碱金属掺杂导致分子筛晶体内产生较强的碱位,分子筛的活性、再生性和选择性因此增强.Kang等[26]通过计算发现Zn原子同晶置换改性的ZnAPO⁃34分子筛,孔道内质子转移不需要能垒,有利于甲醇分子以H3COH2+的形式吸附于分子筛孔道内.但是,对金属同晶置换改性分子筛的作用机理并没有清晰的认识,尤其是在FAU分子筛体系中的理论研究鲜有报道.
本文采用密度泛函理论方法,研究了过渡金属Zn和碱金属Ca同晶置换FAU分子筛骨架原子的
改性机理,并考察其对含氧化合物甲醇、二甲醚、丙醛的吸附性能,为高性能FAU分子筛吸附剂的设计提供理论依据.
1 计算方法与模型
采用基于密度泛函理论的 VASP计算程序软件包[27,28].交换⁃相关能计算采用广义梯度近似(GGA)的PBE[29]泛函,离子与价电子间的相互作用采用投影缀加波(PAW)[30,31]赝势描述,平面波截断能为400 eV,布里渊区积分网格采用Monkhorst⁃Pack(MP)[32]方法,k点取1×1×1.几何结构优化的自洽精度设为原子受力小于0.5 eV/nm,能量收敛标准为1×10-5eV/atom.
FAU分子筛结构模型从MedeA软件数据库中选取,晶胞参数a=b=c=2.5028 nm.FAU骨架的结构单元为β笼,相邻β笼之间通过六方柱连接,形成由硅氧四面体和铝氧四面体通过氧桥键相连组成的孔道和空腔体系[图1(A)].计算中截取FAU的结构单元β笼结构[图1(B)],β笼结构中包含24 个Al原子、24个Si原子和72个O原子,电荷非零,为多电子体系.通过Bader电荷分析可知,Si,Al,O的Bader电荷分别为0.82,0和8.0|e|,Al原子完全失去外层电子,Si和O原子上有电子局域,电子结构如图1(C)所示.

Fig.1 Calculated structure of FAU zeolite(A),βcage(B)and the isosurface(50 e/nm3)of calculated spin charge densities(C)
醇醚分子吸附能(Eads)的计算如下式:
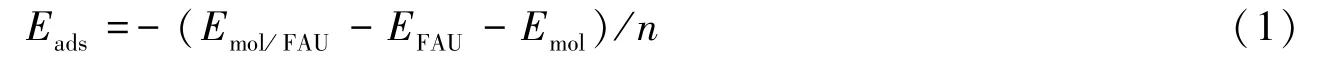
式中,Emol/FAU为FAU的β笼孔道中吸附醇醚分子之后的总能量,EFAU为所截取的β笼的总能量,Emol为醇醚分子的能量,n为吸附醇醚分子的个数,负号保证所计算的吸附能为正值.
2 结果与讨论
2.1 Zn,Ca同晶置换改性分子筛结构
β笼结构是由包含Si—O—Al的八元环和十二元环相互连接组成,具有2种位置不同的Al和Si原子,一种是连接2个八元环的Al和Si原子,一种是连接八元环和十二元环的Al′和Si′原子.Zn和Ca分别取代β笼中的Al和Si原子的优化结构如图2(A)~(D)所示,取代Al′和Si′原子的优化结构如图2(E)~(H)所示.
从图2可见,Zn,Ca同晶置换分子筛骨架原子(Al/Si/Al′/Si′)之后,孔道结构出现不同程度的弛豫,由于Ca原子的半径明显大于Al和Si,所以Ca掺杂的孔道结构弛豫程度最大,而Zn的掺杂对骨架结构影响不大.为了研究在β笼骨架中掺杂Zn,Ca前后的结构变化,利用均方根(RMS)位移公式[33]计算了取代位置(Al/Si/Al′/Si′)的结构弛豫,同时进行了取代能量的计算.
均方根位移(rRMS)的计算如下式所示:

式中,r0i和ri分别为未掺杂结构中Al/Si—O键键长和掺杂结构中Zn/Ca—O键键长.取代能(Esub)的计算如下式所示:
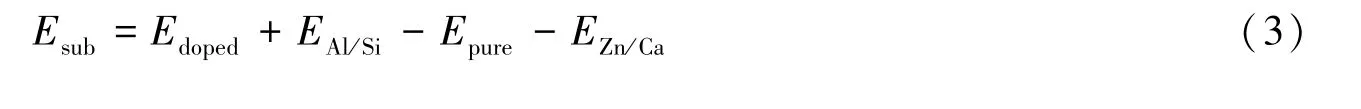

Fig.2 Calculated structures of A l(A,B),Si(C,D),A l′(E,F)and Si′(G,H)atoms substituted byZn(A,C,E,G)and Ca(B,D,F,H)atoms
式中,Edoped为Zn或Ca原子取代β笼骨架中的Al或Si之后结构的总能量,Epure为纯净β笼的能量,EAl/Si和EZn/Ca为Al/Si/Zn/Ca单个原子的能量.
根据式(2)和式(3)计算得到的rRMS和Esub列于表1.从表1可见,Zn掺杂取代的结构弛豫明显小于Ca掺杂,而Zn,Ca掺杂取代的取代能则相反,表明结构弛豫的能量降低了掺杂结构的总能量.所有取代能均为正值,表明Zn,Ca掺杂取代FAU分子筛骨架原子需要消耗能量.Zn,Ca原子取代Al(Al′)原子的取代能分别为4.89 eV(5.05 eV)及0.92 eV(1.00 eV),小于取代Si(Si′)原子的取代能[5.67 eV(5.72 eV)及1.86 eV(1.90 eV)],同时Al和Si原子位置的取代能也略小于Al′和Si′原子位置,表明连接2个八元环骨架原子的Al更容易被杂原子(Zn/Ca)取代,因此选择Zn,Ca掺杂替换Al作为同晶置换改性模型结构.
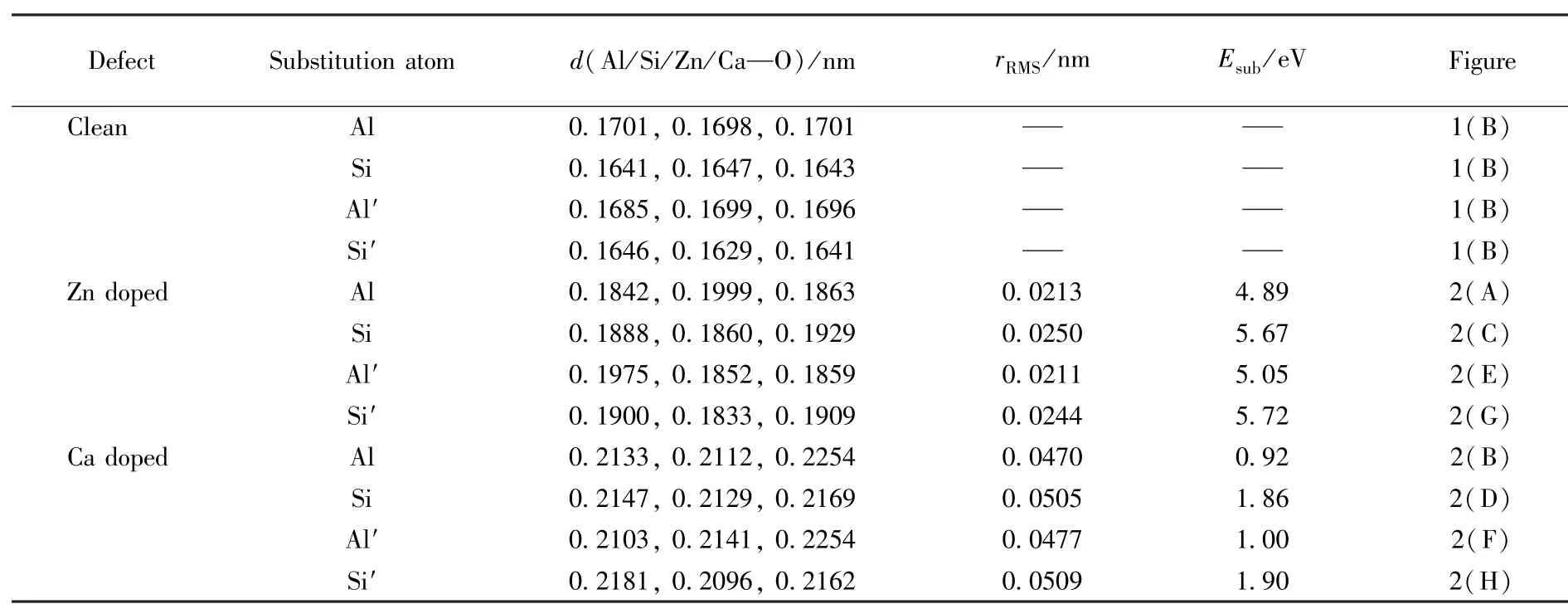
Table 1 Calcu lated lengths of A l/Si/Zn/Ca⁃O bonds,rRMSof different substituted atom s and substitution energies of Zn/Ca dopedβcage
2.2 纯净β笼孔道内甲醇、二甲醚和丙醛的吸附
含氧化合物甲醇、二甲醚和丙醛均为极性分子,而氧原子的电负性较强,电子局域在氧原子上,因此,优化计算了3种含氧化合物在Al和Si 2种金属吸附位上的吸附.计算结果表明,二甲醚和丙醛分子不能吸附于Si原子位,并且在静态优化过程中会自发的转移吸附到邻近的Al原子位上,而甲醇分子在两种金属活性位上都能吸附.
甲醇、二甲醚和丙醛分子在纯净β笼孔道内Al原子上的吸附结构如图3(A)~(C)所示,从图中
可以看出,3种含氧化合物均能稳定吸附于孔道结构内,使孔道产生不同程度的结构弛豫.利用均方根位移公式分析计算了活性位点吸附前后的结构变化(表2),结果表明,甲醇、二甲醚、丙醛分子的吸附对孔道结构的弛豫影响依次增大.从差分电荷的分析[图3(D)~(F)]可以看出,3种含氧化合物与孔道结构之间均存在明显的电荷转移,醇醚分子化学吸附于孔道内.

Fig.3 Calcu lated structures ofm ethanol(A),dimethyl ether(B)and propionaldehyde adsorption(C)at A l site ofβcage and isosurfaces(6 e/nm3)(D—F)of charge redistribution of(A—C),respectively
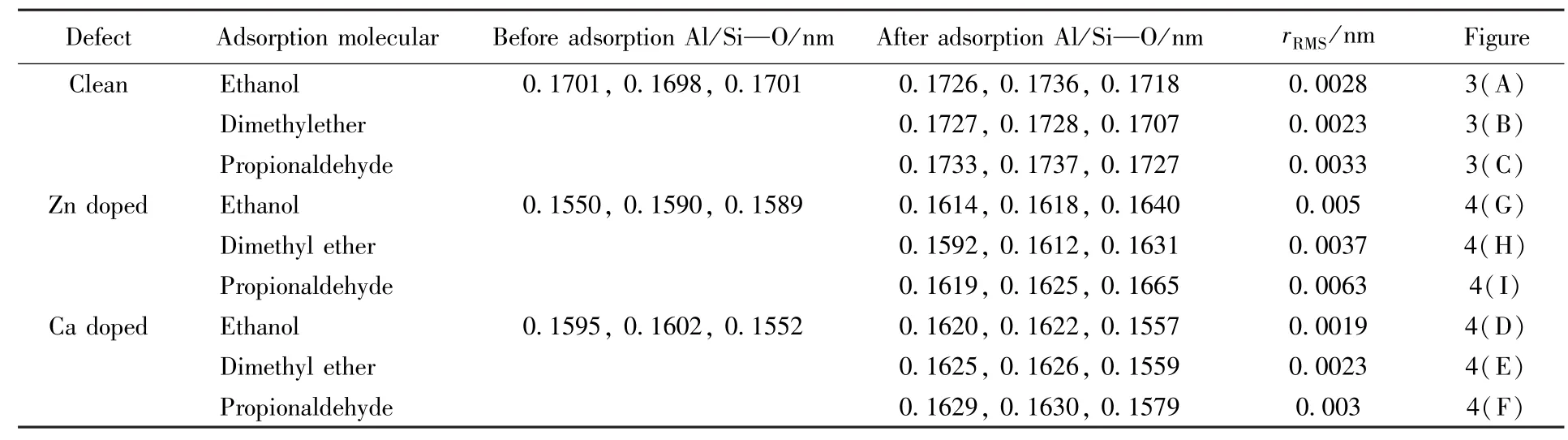
Table 2 Calculated lengths of Al/Si—O bonds,rRMSofmethanol,dim ethyl ether and propionaldehyde adsorption at Al/Si site ofβcage
利用式(1)计算了甲醇、二甲醚及丙醛分子的吸附能,计算结果列于表3.从表3可以看出,甲醇、二甲醚及丙醛分子在Al原子位置都是比较稳定的化学吸附,吸附能分别为0.83,0.97和0.76 eV,在Si原子上甲醇则为较弱的物理吸附(0.54 eV).因此,3种含氧化合物更趋于吸附在Al原子位置.

Table 3 Calculated adsorption energies of ethanol,dim ethyl ether and propionaldehyde at Al and Si sites
2.3 Zn,Ca掺杂孔道内甲醇、二甲醚和丙醛的吸附
以Zn,Ca同晶置换Al原子的掺杂结构为模型,优化计算了甲醇、二甲醚和丙醛在β笼掺杂孔道内的吸附,包括杂原子Zn,Ca吸附位及邻近的Si原子吸附位.计算结果表明,在杂原子Zn位点甲醇、二甲醚和丙醛不能吸附,而在Ca原子位它们的吸附也是非常弱的物理吸附.但是在与Zn或者Ca邻近的Si原子上,3种含氧化合物则能稳定吸附,明显区别于纯净β笼内Si原子完全没有吸附活性的情况.图4为Ca原子和邻近Si原子[Si(Ca),Si(Zn)]上甲醇、二甲醚及丙醛的稳定吸附结构,插图所示
为相应的差分电荷结构,可以看出,3种含氧化合物与孔道结构之间具有明显的电荷转移.

Fig.4 Calculated structures of ethanol(A,D,G),dimethyl ether(B,E,H)and propionaldehyde(C,F,I)adsorption in Ca⁃doped(D—F)and Zn⁃doped(G—I)βcage
利用式(1)计算得到了3种含氧化合物在掺杂β笼孔道内的吸附能(表4).可见,甲醇、二甲醚及丙醛在Zn原子位不吸附,而在Ca原子位吸附,且甲醇的吸附能最大,但只有0.45 eV,属于较弱的物理吸附,表明Zn和Ca的掺杂并没有直接提供给3种含氧化合物活性吸附位.但是由于Zn,Ca的掺杂,原本并没有吸附活性的Si原子位点则可以稳定的吸附甲醇、二甲醚和丙醛分子,并且在Zn掺杂的孔道内3种含氧化合物的吸附均强于纯净孔道内Al原子位上的吸附,这与Yang等[24]的实验结果一致,即杂原子Zn的掺入可以改善分子筛孔道结构的性质.在与Zn邻近的Si原子上,甲醇分子可以自发地解离吸附[图4(G)],吸附能达到2.28 eV;丙醛分子也以双齿吸附于Zn,Ca掺杂的β笼孔道内[图4(I),(F)],吸附能分别为3.93和2.29 eV.同时,从表4可以看到,Zn掺杂结构中3种含氧化合物的吸附强度明显强于Ca掺杂的结构,通过吸附位点Si—O键长均方根位移(表2)的计算可知,在Zn掺杂的孔道内吸附导致的结构弛豫降低了吸附结构的总能量.

Table 4 Calcu lated adsorption energies of ethanol,dimethyl ether and propionaldehydein Zn⁃and Ca⁃dopedβcage
2.4 电子结构分析
为了进一步理解Zn,Ca掺杂对于β笼孔道性质的影响作用,对掺杂孔道结构进行了Bader电荷分
析.截取纯净的、Zn和Ca掺杂的β笼孔道的局部结构,如图5所示.利用Bader电荷分析计算得到了掺杂原子Zn,Ca邻近的Si原子电子结构的变化(表5).从Bader电荷分析的结果可知,与纯净β笼孔道内的Si原子相比,Zn,Ca的掺杂明显改变了邻近Si原子的电子结构,出现了缺电子的空穴,这主要是因为+3价的Al被+2价的Zn和Ca取代之后,孔道骨架结构丢失了1个电子.其中,Ca掺杂的结构2号Si原子完全失掉1个电子,从0.82减小到0;Zn掺杂的结构则是1号Si原子完全失掉1个电子,从0.83减小到0,2种位置的Si原子都具有甲醇、二甲醚及丙醛分子的吸附活性.Li等[34]研究发现,表面局域电子的位置将显著影响表面结构性质,因此,缺电子空穴的位置及其对分子筛孔道结构性质、含氧化合物吸附的影响作用还需进一步的分析研究,以更深入地理解同晶置换改性的机理.电子结构分析结果表明,Zn,Ca的掺杂导致缺电子空穴的产生,使孔道结构局部极性增强,增强了极性分子甲醇、二甲醚和丙醛的吸附.
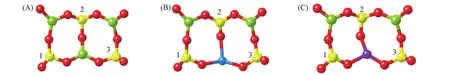
Fig.5 Local structures of clean(A),Ca⁃doped(B),Zn⁃doped(C)βcage

Table 5 Calculated Bader charge of different Si sites
3 结 论
利用密度泛函理论方法计算了纯净的及Zn,Ca掺杂的FAU型分子筛β笼孔道结构内甲醇、二甲醚及丙醛的吸附,并对Zn,Ca掺杂改性的机理进行了计算研究.结果表明,纯净β笼孔道内只有Al原子位点为甲醇、二甲醚和丙醛分子的活性吸附位,Si原子没有吸附活性;而在Zn,Ca掺杂的β笼孔道结构内,杂原子Zn,Ca虽然没有直接提供活性吸附位,但是掺杂导致邻近Si原子位置缺电子空穴的产生,增强了极性分子甲醇、二甲醚及丙醛的吸附.
[1] Xia S.Q.,Wang P.F.,Xu H.S.,Hu J.,Lv D.Q.,Fine Chem.,2014,31(12),1476—1479(夏思奇,王鹏飞,徐华胜,胡杰,吕待清.精细化工,2014,31(12),1476—1479)
[2] Luiz K.C.,Juliana J.R.,Jose R.Z.,Geraldo N.,Cristiano M.B.,Romulo S.A.,Carlos E.F.,Powder Techn.,2012,229,1—6
[3] Sheng X.L.,Zhou Y.M.,Duan Y.Z.,Xue M.W.,J.PorousMat.,2011,18(6),677—683
[4] Yan B.,Mahmood A.,Liang Y.,Xu B.Q.,Catal.Today,2016,269(1),65—73
[5] Zhang Y.Q.,Zheng H.Y.,Zhang R.G.,Li Z.,Wang B.J.,Zhao Q.Y.,Chem.J.Chinese Universities,2015,36(10),1945—1953(张艳青,郑华艳,章日光,李忠,王宝俊,赵秋勇.高等学校化学学报,2015,36(10),1945—1953)
[6] Popovych N.O.,Kyriienko P.I.,Soloviev S.O.,Orlyk S.M.,Dzwigaj S.,Microp.Mesop.Mater.,2016,226(15),10—18
[7] Najar H.,Zina M.S.,Ghorbel A.,React Kinet.Mech.Catal.,2010,100(2),385—398
[8] Lim W.T.,Seo S.M.,Lee O.S.,Wang L.Z.,Lu G.Q.,J.Incl.Phenom.Macrocycl.Chem.,2010,67(3),261—269
[9] Li H.C.,Zhou D.H.,Tian D.X.,ShiC.,Muller U.,Feyen M.,Yilmaz B.,Gies H.,Xiao F.S.,de Vos D.,ChemPhysChem,2014,15(8),1700—1707
[10] Meeprasert J.,Jungsuttiwong S.,Namuangruk S.,Microp.Mesop.Mater.,2013,175,99—106
[11] Ma J.,Qiang L.S.,Wang J.F.,Tang X.B.,Tang D.Y.,J.PorousMat.,2011,18(5),607—614
[12] Wang Q.,Wu Y.J.,Wang J.,Lin X.,Acta Phys.Chim.Sin.,2012,28(9),2108—2114
[13] Hernandez⁃Morales V.,Nava R.,Acosta⁃Silva Y.J.,Pawelec B.,Microp.Mesop.Mater.,2012,160,133—142
[14] Tao L.,Li G.S.,Yin S.F.,Au C.T.,React Kinet.Mech.Catal.,2011,103(1),191—207
[15] Zhang R.Z.,Wang C.,Xing P.,Wen S.B.,Wang J.,Zhao L.F.,Li Y.P.,Chem.J.Chinese Universities,2015,36(4),725—732(张瑞珍,王翠,邢普,温少波,王剑,赵亮富,李玉平.高等学校化学学报,2015,36(4),725—732)
[16] Su X.F.,Wang G.L.,Bai X.F.,Wu W.,Xiao L.F.,Fang Y.J.,Zhang J.W.,Chem.Eng.J.,2016,293,365—375
[17] Jin Y.J.,Asaoka S.,Zhang S.D.,Li P.,Zhao S.L.,Fuel Proc.Techn.,2013,115,34—41
[18] Omata K.,Yamazaki Y.,Watanabe Y.,Kodama K.,Yamada M.,Ind.Eng.Chem.Res.,2009,48(13),6256—6261
[19] Wang Y.,Wu W.,Li C.,Yang J.,Zhou Y.J.,Acta Petrolei Sin.,2011,27(5),682—686(王瑜,吴伟,李程,杨杰,周亚静.石油学报,2011,27(5),682—686)
[20] Geraldo E.,Stevie H.L.,Ana C.R.,Antonio S.A.,Valter J.,J.Mater.Sci.,2010,45(4),1117—1122
[21] Shamzhy M.,Shvets O.,Opanasenko M.,Cejka J.,J.Mater.Chem.,2012,22(31),15793—15803
[22] Ahmed S.,J.Porous Mat.,2012,19(1),111—117
[23] Wang J.H.,Xie J.Y.,Zhou Y.,Wang J.,Microp.Mesop.Mater.,2013,171,87—93
[24] Yang C.G.,Qiu M.H.,Hu S.W.,Chen X.Q.,Zeng G.F.,Liu Z.Y.,Sun Y.H.,Microp.Mesop.Mater.,2016,231,110—116
[25] Zhou Y.,Jin Y.H.,Wang M.,Zhang W.,Xie J.Y.,Gu J.,Wen H.M.,Wang J.,Peng L.M.,Chem.Eur.J.,2015,21(43),15412—15420
[26] Kang L.H.,Zhang T.,Liu Z.M.,Han K.L.,J.Phys.Chem.C,2008,112(14),5526—5532
[27] Kresse G.,Furthmüller J.,Comp.Mater.Sci.,1996,6,15—50
[28] Kresse G.,Furthmüller J.,Phys.Rev.B,1996,54(16),11169—11186
[29] Perdew J.P.,Burke K.,Ernzerhof M.,Phys.Rev.Lett.,1996,77(18),3865—3868
[30] Kresse G.,Joubert D.,Phys.Rev.B,1999,59(3),1758—1775
[31] Blöchl P.E.,Phys.Rev.B,1994,50(24),17953—17979
[32] Monkhorst H.J.,Pack J.D.,Phys.Rev.B,1976,13,5188—5192
[33] Wang H.F.,Gong X.Q.,Guo Y.L.,Guo Y.,Lu G.Z.,Hu P.,J.Phys.Chem.C,2009,113(23),10229—10232
[34] Li H.Y.,Wang H.F.,Gong X.Q.,Guo Y.L.,Guo Y.,Lu G.Z.,Hu P.,Phys.Rev.B,2009,79(19),193401—193405
Theoretical Studies on the Structure and Adsorption Properties of Isomorphously Substituted FAU Zeolite†
YANG Bingxing1,2,YE Liping1,2∗,GU Huijie1,2,XU Huasheng2,3,LUO Yong1,2,LIHuiying4
(1.State Key Laboratory ofPolyolefins and Catalysis,Shanghai200062,China;2.Shanghai Research Institute ofChemical Industry,Shanghai200062,China;3.Shanghai Lüqiang New Materials Co.,Ltd.,Shanghai201608,China;4.Research Institute ofApplied Catalysis,Shanghai Institute of Technology,Shanghai201418,China)
The adsorption of oxygenated chemicals(methanol,dimethyl ether,propionaldehyde)in channel structure of FAU type zeoliteβcage,aswell as themodificationmechanism of Zn/Ca isomorphous substitution were investigated bymeans of density functional theory(DFT)calculations.The results indicate thatmethanol,dimethyl ether and propionaldehyde can chemisorb at the top site of the Al atom,while they can barely adsorb at the Si atom.For Zn⁃/Ca⁃dopedβcage,Al3+was substituted by Zn2+/Ca2+,a hole was formed on the adjacent Si atom due to the loss of one electron,therefore the adsorption of methanol,dimethyl ether and propionaldehyde were enhanced.However,the dopant atom itself doesn't serve as adsorption site.
Density functional theory(DFT);FAU zeolite;Doping;Adsorption
O641
A
10.7503/cjcu20160449
(Ed.:Y,Z,S)
†Supported by the Shanghai Zhangjiang National Innovation Demonstartion Zone Key Projects of Special Development Funds,China(No. 201505⁃PT⁃B108⁃004).
2016⁃06⁃22.
日期:2016⁃10⁃18.
上海张江国家自主创新示范区专项发展资金重点项目(批准号:201505⁃PT⁃B108⁃004)资助.
联系人简介:叶丽萍,女,博士,高级工程师,主要从事催化新材料研究.E⁃mail:ylp_by@126.com
