H1N1流感病毒的HA、NA蛋白序列进化树
2016-11-21李建林薛晓丽唐旭清
李建林,薛晓丽,唐旭清
(江南大学 理学院,江苏 无锡214122)
H1N1流感病毒的HA、NA蛋白序列进化树
李建林,薛晓丽,唐旭清*
(江南大学 理学院,江苏 无锡214122)
为了从本质上揭示H1N1病毒分子的变异、流感流行等关系,提出一种构建H1N1型流感病毒进化树的新方法。在1902—2013年全球22 455条H1N1型流感病毒HA蛋白序列和16 444条NA蛋白序列数据的基础上,利用其特征向量构建基于内积的蛋白序列相似度;采用基于相似度的完全聚类图的方法进行数据系统粗粒化的相似信息提取;最后,利用基于模糊邻近关系的结构聚类方法构建H1N1型禽流感病毒HA、NA蛋白序列的进化树及算法研究。试验结果表明:H1N1病毒的变异不仅与爆发时间密切相关,还与所分布地域及地域间的距离有很大关系,且分布地域间的距离越近,爆发的病毒进化的相似程度越高。因此这种基于大数据处理的新方法能有效揭示流感病毒的进化关系,为进一步研究流感病毒的变异、进化与预测奠定了基础。
H1N1病毒;HA蛋白序列;NA蛋白序列;模糊邻近关系;结构聚类;进化树
自2009年全球出现了可以直接感染人的流感病毒H1N1后,流感对人类的威胁已经不可忽视[1]。流感病毒是一种单负链的RNA病毒,有8个负链的RNA单链片段组成。这8个片段共编码10种病毒蛋白质,即结构蛋白质(HA、NA、NP、M1、M2、PBl、PB2、PA)和非结构蛋白质(NSl、NS2)。依据位于病毒外膜的血凝素(HA)和神经氨酸酶(NA)蛋白抗原性的不同,目前可将禽流感病毒分为16个H亚型(Hl—H16)和10个N亚型(N1—N10)。HA是流感病毒表面的主要蛋白质之一,与流感的发生和流行最为密切,是病毒抗原性变异的分子基础[2]。因此,对流感HA蛋白序列分类,可以从本质上揭示流感病毒分子结构的变异,为预防、控制流感发生提供理论依据[3-4]。NA作为H1N1流感病毒表面的主要蛋白质,也是H1N1病毒的主要抗原之一,在流感病毒的传播中起着非常重要的作用[5]。对流感病毒NA蛋白序列的同源性研究分析,也是目前研究流感病毒变异进化的重要手段之一[6-8]。
蛋白质的进化树利用分支层次或拓扑图形直观地体现了蛋白质的进化过程,它是产生新的基因复制或享有共同祖先的蛋白质的歧异点的一种反映[9-10]。蔡斌[11]等基于氨基酸序列的进化距离,对不同爆发点的H5N1型禽流感进行进化树聚类,将病毒株进行分类,研究其序列变异特点。郝荣超[12]等基于模糊聚类对中国荷斯坦牛和鲁西黄牛的DNA D-loop区部分序列进行分析,构建进化树,研究它们的群体遗传性以及起源进化。基于模糊邻近关系的结构聚类[13-15]是构建蛋白序列进化树的一种有效方法。但是考虑到算法的复杂度,该方法只适用于对小型数据进行分析。 对于大型的数据,需要通过数据挖掘手段对其进行粗粒化(或模块化)提取[16],压缩成小型数据,再用该方法构建进化树。
在1902—2013年全球爆发的H1N1型禽流感病毒的22 455条HA蛋白序列和16 444条NA蛋白序列数据基础上,利用其特征向量[17]构建基于内积的蛋白序列相似度。采用基于相似度的完全聚类图[18-19]方法进行数据系统粗粒化的相似信息提取。最后,利用基于模糊邻近关系的结构聚类[13,15]方法进行H1N1型禽流感病毒的进化树研究。旨在从本质上揭示H1N1病毒分子的变异、流感流行等关系,为预防、控制和预报流感发生提供基础理论依据。
1 数据来源与方法
1.1 数据来源
NCBI是美国的一个大型生物信息学系统,主要通过NCBI网站 (http://www.ncbi.nlm.nih.gov/)为全世界的科学家服务,它拥有很多数据库查询工具,主要包括:GenBank,Molecular Databases和Literature Databases。 从 NCBI网站中 Molecular Databases的Protein Sequence上下载了1902—2013年H1N1病毒的22 455条HA蛋白序列和16 444条NA蛋白序列。在数据基础上开展数据实验工作。
1.2 方法
1.2.1 生成特征向量 从氨基酸序列预测蛋白质的结构和亚细胞位置,不仅是生物信息学科的重要研究领域,也是多类模式识别问题,而在模式识别问题中,特征选取和描述是决定分类质量的关键[20]。现有的氨基酸序列特征描述方法主要有两类:单纯基于氨基酸残基序列的方法和考虑氨基酸性质的描述方法。本文中运用了一种改进的基于疏水模式的氨基酸性质描述方法[17],构建特征向量。
根据氨基酸的亲疏水性,将20种氨基酸分为四大类:强亲水、强疏水、弱亲水或弱疏水、无明显性质,分别用qq、qs、r和w来表示,即
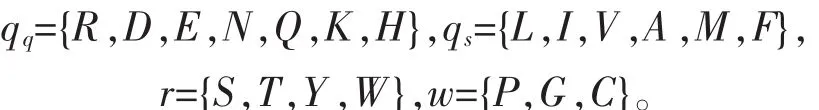
对于一个给定长为n的蛋白序列S=s1,s2,L,sn,其中si(i=1,2,……,n)是20种氨基酸中的一种,定义

则得到一条序列X (s)=c1,c2,……,cn,ci∈{L,B,W,P}。记an是长度为n的蛋白序列中a类氨基酸出现的总数,其中a代表L、B、W和P任意一种;记bn为简化前序列中氨基酸的总数,其中代表20种氨基酸中的任意一种;对分类后的4组氨基酸考虑其二肽结构,即它们的两两组合:LL、LB、LW、LP、BL、BB、BW、BP、WL、WB、WW、WP、PL、PB、PW、PP。记cn为分类后序列中出现每一类二肽的总数,其中c表示16种二肽中的任意一种。分别计算a、b、c的相对频率fa,gb和hc如下:
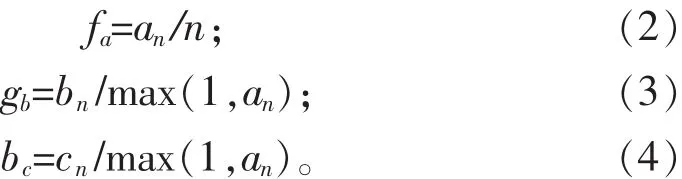
这样,就把长度为n的蛋白序列映射成一个40维向量V(C)。
1.2.2 基于内积的相似度 对于两个蛋白序列i和j,由内积可定义它们之间的相似度如下:
其中V(i)和V(j)分别代表i和j对应的40维向量。显然基于内积的相似度是一个可分离的模糊邻近关系[13]。
1.2.3 基于相似度的完全图聚类方法 聚类分析方法包括系统聚类法、分解法、加入法、动态聚类法等,而最常用的方法是系统聚类法,也称结构聚类方法或层次聚类分析方法[15]。但是在大数据的挖掘过程中,传统的聚类方法还是存在一些问题,如:模糊均值聚类[21-23]是目前为止应用最广且极有影响的聚类方法,该算法可以将确定的k个划分达到平方误差最小,对于处理大数据集,该算法是相对可伸缩和高效的。由于算法中的k是事先给定且初始聚类中心的选择对聚类结果影响很大,当数据量非常大时,不利于数据的分析[23]。为了解决这些问题,本文结合已有的完全图聚类模型[18-19],基于相似度对数据集进行完全图聚类。具体过程如下:
1)设定一个聚类阈值λ及聚类数k=1。
2)从数据集中任意选取两个初始对象i和j,按照上述给定的相似度计算两个初始对象间的相似度sij。若sij>λ,则对象i和j之间构成完全图,将它们归为一类;否则,二者分为不同类。此时k⇐k+1。
3)从数据集中引入第n个对象,计算其与之前n-1对象所组成的k类中每一类的所有对象的相似度。如果该对象与第m(m≤k)类中所有元素之间的相似度都大于阈值λ,说明该对象与第m类中所有的对象构成完全图,将其归入第m类中;否则,说明它自身构成完全图,将其另归一类。此时k⇐k+1。
4)重复过程3),直到数据集中所有的对象均被引入。输出,算法结束。
利用Matlab应用软件编程,具体算法A如下:
输入:阈值λ,病毒集A={a1,a2,……,aN},初始聚类数k=0
输出:聚类结果和最终聚类数k

2 数据处理
2.1 数据初步处理
首先,挑选数据集。网站内的数据由测序数据及论文索引数据构成,不排除一些由于主观因素而导致的异常数据。如 1993年的 A/Wilson-Smith/ 1933(H1N1)的HA序列长度异常,不利于统计研究,因此需将此类异常数据剔除,同时也将对应的NA蛋白序列剔除;由于NCBI网站中数据的不完整性:HA蛋白序列有22 455个,而NA蛋白序列只有16 444个,不能达到匹配。通过Matlab编程识别同种病毒的HA和NA蛋白序列,将其挑选出来,以年为单位分别放到不同的文件夹中。
其次,以年为单位,利用特征向量计算公式计算出H1N1病毒i的HA蛋白序列H(i)和NA蛋白序列对应的特征向量N(i),取平均值代表H1N1病毒特征向量V(i),即V(i)=[H(i)+N(i)]/2,利用算法A对每年的病毒聚类。
第三,计算每一类病毒的几何中心,选出该类中与之相似度最大的病毒作为该类的代表病毒,被选出的病毒可以代表其所属类别的所有属性。
最后,将上述步骤选出来的所有代表病毒放入同一个文件夹中存储。重复使用算法A,对选出的病毒集进行二次聚类,选出代表病毒,这样就对数据进行了全局的粗粒化提取。
取λ1=0.999时,利用算法A对数据进行局部聚类,筛选出了67个不同年份、不同地区的H1N1病毒。这些病毒包含了数据集中来自所有年份的病毒所带有的信息;在进行全局聚类时,取λ2=0.999 5,得到了23个H1N1病毒。为了方便叙述,给23个病毒编号,如表1所示。

表1 筛选的23个H1N1病毒与相应的编号Table 1 Filtered 23 H1N1 viruses and their corresponding serial numbers
2.2 基于模糊邻近关系构建进化树
利用相似度公式计算提取出的所有病毒之间的相似度,得到一个对称矩阵。基于模糊邻近关系的结构聚类方法,利用文献[13]中的算法B对23个代表病毒之间的相似度矩阵进行聚类,得到进化树,如图1所示。

图1 筛选的23个H1N1病毒组成的进化树Fig.1 Evolutionary tree of the filtered 23 H1N1 viruses
3 结果与讨论
从病毒信息很容易看出:有些年份 (如2012年)的病毒并没有被选出来作为代表,这说明该年份的所有病毒与其他年份病毒属性相似,可以归到其他年份所代表的病毒之中。以经过局部聚类所选出来的病毒 A/swine/Indiana/2012(H1N1)和 A/ Ulaanbaatar/2011(H1N1)为例,通过全局聚类,这两类病毒均与A/Karasuk/2010(H1N1)有高于0.999 806的相似度,最终被归入A/Karasuk/2010(H1N1)所代表的病毒类中。
从系统进化树上可以明显看出,同一地点出现的病毒,如10、11所代表的A/swine/Wisconsin/1961(H1N1)和A/swine/Wisconsin/1970(H1N1)的蛋白序列之间相似度很高,很早就聚成一类。说明它们之间一定存在着某种进化关系,且后者是由前者发生基因重组和变异得来的。
从进化树中还可以直观地看到(7,12,15,22),(5,9,8,13,10,11,16,20,6,4)和(14,19,18,21)形成了相对独立的3个分支。这些分支中大部分的病毒的发生时间是相近的,如分支(14,19,18,21)中的病毒有3个是爆发在1996—2003这几年间,这说明了发生时间相近的病毒在进化过程中有密切关系。而每个分支中也有相对年份比较早的病毒,如(5,9,8,13,10,11,16,20,6,4)中的 4和 5代表的病毒分别是1934年和1940年爆发的病毒,而其他病毒均与其爆发时间相距较远,但它们与4和5之间相似度却很高,可以推断这一分支中的其他病毒均由病毒4和5变异而来。而纵观全局,所有的病毒最终都是从由1所代表的1918年爆发的那一类病毒进化而来的。
此外,在爆发时间相近或相同的情况下,同一地域的病毒也分成了小的分支。如(7,12,15,22)中的所有病毒发生地均为北美洲;(5,9,8,13,10,11,16,20,6,4)中 (4,16,20)发生地是亚洲,(10,11)发生地是美洲,(8,9)发生地是欧洲;(14,19,18,21)中分支(14,19)的发生地是西欧,分支(18,19)发生地是亚洲,这充分说明病毒的进化过程与所分布地域及地域间的距离有很大关系,即分布地域间的距离越近,爆发的病毒进化的相似程度越高,这一结果与文献[24-26]一致。同时,在地域与爆发时间相近的情况下,如 (14,19)和(4,16)也形成各自的分支,且它们都有同一类型的宿主,此表明病毒的进化与宿主具有一定的关系。这些结果与文献[24-26]相吻合。
在图1中还能观察到:一些病毒爆发时间、地域、宿主都不相同,如(4,13),但是却在同一分支上。 这可能是因为一些特殊因素,如候鸟迁徙、人工养殖等所致[26]。
4 结语
通过对22 455个H1N1病毒HA蛋白序列和16 444个NA蛋白序列进行信息筛选,选出了23个代表病毒,对病毒进行聚类分析,得到系统进化树,反映了各类病毒之间的进化关系。试验结果分析表明:H1N1病毒的变异与地域及地域间的距离、爆发时间等有密切关系。这一结果与已有的研究结果相吻合。因此,这种基于大数据处理的新方法能有效揭示流感病毒的进化关系,为进一步研究流感病毒的变异、进化与预测奠定了基础。
[1]SIMTH G J D,VIJAYKRISHNA P V,BAHL J,et al.Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic[J].Nature,2009,459(7250):1122-1125.
[2]于虹,温晶,杨裔,等.甲型流感病毒H1N1 HA蛋白在果蝇S2细胞中的表达及免疫原性研究 [J].中国人兽共患学报,2012,28(9):875-879. YU Hong,WEN Jing,YANG Yi,et al.Expression of influenza virus A(H1N1)hemagglutinin(HA)in drosophila S2 cell lines and its immunogenicity analysis in BALB/c mice[J].Chinese Journal of Zoonoses,2012,28(9):875-879.(in Chinese)
[3]颜健华,梁丹洁,李春英,等.H1N1猪流感病毒广西分离株HA基因序列分析[J].南方农业学报,2013,44(7):1196-1200. YAN Jianhua,LIANG Danjie,LI Chunying,et al.Analysis of HA gene sequence of a subtype H1N1 swine influenza virus isolated from Guangxi strains[J].Journal of Southern Agriculture,2013,44(7):1196-1200.(in Chinese)
[4]王勇,薛颖,陈淑霞,等.H3N2亚型人流行性感冒病毒HA1的蛋白序列同源性比较、变异规律及结构与功能的分析[J].病毒学报,2002,18(4):289-296. WANG Yong,XUE Ying,CHEN Shuxia,et al.Analysis and studies on the amino acid sequence homology,mutant regularity,structural and functional relationship of HA1 of human influenza virus(H3N2)[J].Chinese Journal of Virology,2002,18(4):289-296.(in Chinese)
[5]许沙沙,常彦敏,徐霖,等.2010年广州市甲型H1 N1流感病毒分离株NA基因变异分析[J].检验医学,2014,29(1):42-49. XU Shasha,CHANG Yanmin,XU Lin,et al.Analysis on neuraminidase(NA)gene variation in imported influenza A(H1 N1)virus from Guangzhou in 2010[J].Laboratory Medicine,2014,29(1):42-49.(in Chinese)
[6]INOUE E,OSAWA Y,OKAZAKI K,et al.An NA-deficient 2009 pandemic H1N1 influenza virus mutant can efficiently replicate in cultured cells[J].Archives of Virology,2014,159(4):797-800.
[7]黄吉城,钟玉清,施永霞,等.输入性甲型H1N1流感病毒神经氨酸酶(NA)基因变异分析[J].中国国境卫生检疫杂志,2014,3(2):73-76. HUANG Jicheng,ZHONG Yuqing,SHI Yongxia,et al.Analysis on the NA gene variation of influenza A (H1 N1)virus[J]. Chinese Frontier Health Quarantine,2014,3(2):73-76.(in Chinese)
[8]田疆,周经娇,陈艺韵,等.甲型H1N1流感病毒神经氨酸酶基因遗传进化分析[J].中山大学学报,2012,31(2):207-212. TIANG Jiang,ZHOU Jinjiao,CHEN Yiyun,et al.Genetic evolution of neuraminidase gene of influenza A/H1N1 virus[J]. Journal of Sun Yat-Sen University,2012,31(2):207-212.(in Chinese)
[9]QI X,CHANDERBALI A S,WONG G K,et al.Phylogeny and evolutionary history of glycogen synthase kinase 3/SHAGGY-like kinase genes in land plants[J].BMC Evolutionary Biology,2013,13(13):143.
[10]骆嘉伟,殷志强,刘淑燕.一种新的关联特征和模糊聚类的进化树构建方法[J].计算机应用研究,2011,28(8):2844-2847. LUO Jiawei,YIN Zhiqiang,LIU Shuyan.Novel method for phylogenetic tree construction based on correlation feature and fuzzy clustering[J].Application Research of Computers,2011,28(8):2844-2847.(in Chinese)
[11]蔡斌,彭谨,江华.基于基因进化树和地理数据库追踪禽流感病毒变异[J].中华急诊医学杂志,2012,21(8):887-891. CAI Bin,PENG Jin,JIANG Hua.Tracking the spread of avian influenza in China:a model based on evolutionary genetics analysis and geographic visualization[J].Chinese Journal of Emergency Medicine,2012,21(8):887-891.(in Chinese)
[12]郝荣超,王国华,常玉霞,等.中国荷斯坦牛和鲁西黄牛mtDNA D-loop序列多态性分析 [J].基因组学与应用生物学,2011,30(5):544-549. HAO Rongchao,WANG Guohua,CHANG Yuxia,et al.Sequence diversity of mitochondrial DNA D-loop region in the population of chinese holstein cattle and Luxi cattle[J].Genomics and Applied Biology,2011,30(5):544-549.(in Chinese)
[13]TANG X Q,ZHU P.Hierarchical clustering problems and analysis of fuzzy proximity relation on granular space[J].IEEE Transactions on Fuzzy Systems,2013,21(5):814-824.
[14]TANG X Q,ZHU P,CHENG J X.The structural clustering and analysis of metric based on granular space[J].Pattern Recognition,2010,43(11):3768-3786.
[15]唐旭清,方雪松,朱平.基于模糊邻近关系的结构聚类[J].系统工程理论与践,2010,30(11):1986-1996. TANG Xuqing,FANG Xuesong,ZHU Ping.Structural clusters based on fuzzy proximity relations[J].Systems Engineering-Theory&Practice,2010,30(11):1986-1996.(in Chinese)
[16]梅娟,何胜,王正祥,等.基于网络模块性的蛋白质序列聚类[J].食品与生物技术学报,2010,29(1):123-127. MEI Juan,HE Sheng,WANG Zhengxiang,et al.Clustering protein sequences through modularity optimization[J].Journal of Food Science and Biotechnology,2010,29(1):123-127.(in Chinese)
[17]钱盼盼.蛋白质序列新的表示方法[D].威海:山东大学威海校区,2011.
[18]DJIDJEV H N,ONUS M.Scalable and accurate graph clustering and community structure detection[J].IEEE Transaction on Parallel and Distributed Systems,2013,24(5):1022-1029.
[19]周翔翔,姚佩阳,王欣,等.基于图论的作战指挥决策群组划分算法[J].系统工程与电子技术,2011,33(3):575-580. ZHOU Xiangxiang,YAO Peiyang,WANG Xin,et al.Algorithm of combat command decision group partition based on graph theory[J].Systems Engineering and Electronics,2011,33(3):575-580.(in Chinese)
[20]靳利霞,唐焕文.氨基酸序列的特征描述[J].计算机与应用化学,2003,20(1):1-5. JING Lixia,TANG Huawen.Characteristic description of amino acid sequences[J].Computers and Applied Chemistry,2003,20(1):1-5.(in Chinese)
[21]CHEN N,XU Z,XIA M.Hierarchical hesitant fuzzy K-means clustering algorithm [J].Applied Mathematics-A Journal of Chinese Universities,2014,29(1):1-17.
[22]周世兵,徐振源,唐旭清.K-means算法最佳聚类数确定方法[J].计算机应用,2010,30(8):1995-1998. ZHOU Shibing,XU Zhenyuan,TANG Xuqing.Method for determining optimal number of clusters in K-means clustering algorithm[J].Journal of Computer Applications,2010,30(8):1995-1998.(in Chinese)
[23]高新波.模糊聚类分析及其应用[M].西安:西安电子科技大学出版社,2004:37-46.
[24]LI J,SHAO T J,YU X F,et al.Molecular evolution of HA gene of the influenza A H1N1 pdm09 strain during the consecutive seasons 2009-2011 in Hangzhou,China:Several immune-escape variants without positively selected sites[J].Journal of Clinical Virology,2012,55(4):363-366.
[25]MULLICK J,CHERIAN S S,POTDAR V A,et al.Evolutionary dynamics of the influenza A pandemic(H1N1)2009 virus with emphasis on Indian isolates:Evidence for adaptive evolution in the HA gene[J].Infect Genet Evol,2011,11(5):997-1005.
[26]程成,张玉稳,柴洪亮,等.一株野鸟源H1N1亚型禽流感病毒的遗传进化特征及其系统学分析 [J].东北林业大学学报,2011,39(1):102-107. CHENG Cheng,ZHANG Yuwen,CHAI Hongliang,et al.Phylogenetic variation in avian influenza virus subtype H1N1 isolated from wild mallard[J].Journal of Northeast Forestry University,2011,39(1):102-107.(in Chinese)
Research on the Evolutionary Tree for H1N1 Flu Virus Based on HA and NA Protein Sequences
LI Jianlin,XUE Xiaoli,TANG Xuqing*
(School of Science,Jiangnan University,Wuxi 214122,China)
The goal of this paper is to propose a new method for constructing evolutionary tree of H1N1 flu viruses in order to reveal the relationship between the molecular variation of H1N1 and epidemics.First,based on the 22455 HA and 16444 NA protein sequence data of H1N1 flu viruses from 1902 to 2013 years,the similarity index of protein sequences was constructed by using inner product of their eigenvectors.Then,the coarse-graining similar information of data was extracted by applying the complete graph clustering based on the similarity index of protein sequences.Finally,the evolutionary tree for HA and NA protein sequences of H1N1 flu viruses was studied by using the structure clustering method based on fuzzy proximity relations.Test results shown that the mutation of the H1N1 viruses was not only closely related to its outbreak time,but also to the outbreak regions and the geographical distances among the distribution regions,and the closer distance between the geographical distribution and the outbreak of the H1N1 viruses,the higher similarity degree of of the H1N1 viruses.Therefore,the new method based on the large data processing can effectively revealthe evolutionary relationship of H1N1 flu viruses,and can provide a foundation for further study of the mutation,evolution and prediction of flu viruses.
H1N1 virus,HA protein sequences,NA protein sequences,fuzzy proximity relations,structural clustering,evolutionary tree
Q 811.4;O 29
A
1673—1689(2016)010—1035—06
2014-10-28
国家自然科学基金项目(11371174);中央高校基本科研业务费专项(JUSRP51317B)。
*通信作者:唐旭清(1963—),男,安徽望江人,工学博士,教授,主要从事智能计算、生态系统建模与仿真、生物信息学研究。E-mail:txq5139@jiangnan.edu.cn
