Phylogeny of Murray Valley encephalitis virus in Australia and Papua New Guinea
2016-11-14EleonoraCellaIvanGabrielliGianguglielmoZehenderMartaGiovanettiAlessandraLoPrestiAlessiaLaiGiordanoDicuonzoSilviaAngelettiMarcoSalemiMassimoCiccozzi
Eleonora Cella, Ivan Gabrielli, Gianguglielmo Zehender, Marta Giovanetti, Alessandra Lo Presti, Alessia Lai, Giordano Dicuonzo, Silvia Angeletti, Marco Salemi, Massimo Ciccozzi,8*
1Department of Infectious, Parasitic and Immunomediated Diseases, National Institute of Health, Rome, Italy
2Department of Public Health and Infectious Diseases, Sapienza University of Rome, Rome, Italy
3University of Rome ‘La Sapienza’
4L. Sacco Department of Biomedical and Clinical Sciences, University of Milan, Italy
5University of Rome ‘Tor Vergata’
6Clinical Pathology and Microbiology Laboratory, University Hospital Campus Bio-Medico of Rome, Italy
7Department of Pathology and Laboratory Medicine, University of Florida, Gainesville, Florida, USA
8University Hospital Campus Bio-Medico, Rome, Italy
Phylogeny of Murray Valley encephalitis virus in Australia and Papua New Guinea
Eleonora Cella1,2, Ivan Gabrielli3, Gianguglielmo Zehender4, Marta Giovanetti1,5, Alessandra Lo Presti1, Alessia Lai4, Giordano Dicuonzo6, Silvia Angeletti6, Marco Salemi7, Massimo Ciccozzi1,8*
1Department of Infectious, Parasitic and Immunomediated Diseases, National Institute of Health, Rome, Italy
2Department of Public Health and Infectious Diseases, Sapienza University of Rome, Rome, Italy
3University of Rome ‘La Sapienza’
4L. Sacco Department of Biomedical and Clinical Sciences, University of Milan, Italy
5University of Rome ‘Tor Vergata’
6Clinical Pathology and Microbiology Laboratory, University Hospital Campus Bio-Medico of Rome, Italy
7Department of Pathology and Laboratory Medicine, University of Florida, Gainesville, Florida, USA
8University Hospital Campus Bio-Medico, Rome, Italy
Accepted 15 March 2016
Available online 20 April 2016
Murray Valley encephalitis virus
Objective: To study the genetic diversity of Murray Valley encephalitis virus (MVEV) in Australia and Papua New Guinea. Methods: MVEV envelope gene sequences were aligned using Clustal X and manual editing was performed with Bioedit. ModelTest v. 3.7 was used to select the simplest evolutionary model that adequately fi tted the sequence data. Maximum likelihood analysis was performed using PhyML. The phylogenetic signal of the dataset was investigated by the likelihood mapping analysis. The Bayesian phylogenetic tree was built using BEAST. Results: The phylogenetic trees showed two main clades. The clade Ⅰincluding eight strains isolated from West Australia. The clade Ⅱ was characterized by at least four epidemic entries, three of which localized in Northern West Australia and one in Papua New Guinea. The estimated mean evolutionary rate value of the MVEV envelope gene was 0.407×10-3substitution/site/year (95% HPD: 0.623×10-4-0.780×10-3). Population dynamics defi nes a relative constant population until the year 2000, when a reduction occurred, probably due to a bottleneck. Conclusions: This study has been useful in supporting the probable connection between climate changes and viral evolution also by the vector point of view;multidisciplinary monitoring studies are important to prevent new viral epidemics inside and outside new endemic areas.
1. Introduction
Murray Valley encephalitis virus (MVEV) is a zoonotic Flavivirus,member of the Japanese encephalitis serocomplex, which is actually endemic in Northern West Australia and Papua New Guinea (PNG). MVEV primarily exists in a transmission cycle between Culex annulirostris and birds. It is considered the causal agent of Murray Valley encephalitis (previously known as Australian encephalitis)and humans are generally considered to be dead-end hosts[1]. The evolution of MVEV in Australia and PNG has proceeded independently and the MVEV strains in circulating Australian are not systematically re-seeded from endemic regions of PNG. Major outbreaks of MVEV occurred in Australia in 1951, 1956 and 1974,with the virus fi rst being isolated during the 1951 outbreak. MVEV may have caused earlier outbreaks of Australian ‘X’ disease in 1917-1918, 1922 and 1925[2]. In the most recent outbreak of MVEV in 1974, 58 cases of encephalitis were identifi ed[3], indicating the signifi cance of this disease despite the infrequency of epidemics. The majority of infections with MVEV are asymptomatic or cause a non-specific febrile illness usually accompanied by headache,myalgia and occasionally rash[6]. However, approximately 1:150 to 1:1 000 infections with MVEV, clinical encephalitis results[4]. After the incubation period of up to four weeks, clinical cases usually present with fever (commonly accompanied by convulsions in children), headache, malaise, and altered mental status, which may be followed by progressive neurological deterioration, parkinsonian tremor, cranial nerve palsies, peripheral neuropathy, coma, fl ac-cid paralysis, and death[4].
Since the mid-1970s a surveillance program for MVEV has been in place in West Australia. Surveillance using sentinel chicken fl ocks is a good system to monitor the spread of infection. In addition,mosquito collection trips are undertaken annually during the latter part of the wet season. Opportunistic mosquito collections are conducted in other regions of Northern West Australia, generally following the detection of MVEV seroconversions in sentinel chickens.Prevention and control of MVEV are still a challenge because include diffi culties in controlling mosquito numbers during periods of extensive fl ooding, and a lack of other prophylactic or treatment measures for MVEV. Mosquitoes are processed for virus isolation to determine infection rates in mosquitoes and to investigate which mosquito species are involved in MVEV transmission[4]. The aim of this study was to define the phylogenic analysis of MVEV in Australia and Papua New Guinea, under a time-scale and a population dynamics, considering the recent GenBank entries not previously subjected to phylogenetic studies[5].
2. Materials and methods
The dataset consisted in 45 sequences of envelope (E) and polyprotein gene, downloaded from GeneBank (http://www.ncbi. nlm.nih.gov/genbank/) and isolated in diff erent regions of Northern West (NW) Australia, except for the two strains isolated in PNG, and selected on the basis of the following criteria: (1) all the available sequences (2) already published in peer-reviewed journals and with(3) known sampling date and location.
All MVEV E gene sequences were aligned using Clustal X and manual editing was performed with Bioedit to a final alignment length of 462 bp[6] after removing gaps. ModelTest version 3.7 was used to select the simplest evolutionary model that adequately fi tted the sequence data[6]. The phylogenetic signal of the dataset was investigated by using of the likelihood mapping analysis of 10 000 random quartets by using TreePuzzle as already described[7]. Groups of four randomly chosen sequences (quartets) were evaluated using Maximum likelihood. For each quartet, the three possible unrooted trees were reconstructed under the selected substitution model. If more than 30% of the dots fall into the center of the triangle, the data are considered unreliable for the purposes of phylogenetic inference. The best-fi tting nucleotide substitution model was tested with a hierarchical likelihood ratiotest implemented in Modeltest software v. 3.7[6]. The statistical robustness and reliability of the branching order within the phylogenetic tree was confi rmed by the approximate likelihood-ratio test approach; all calculations were performed using PhyML[8]. Statistical support for specific clades and clusters was assessed by bootstrap analysis considering bootstrap values>70%. The dated tree, was estimated by using a Bayesian MCMC approach(Beast v. 1.7.4, http://beast.bio.ed.ac.uk)[9] implementing a HKY using both a strict and an uncorrelated log-normal relaxed clock model.
As coalescent priors, we compared three parametric demographic models of population growth (constant size, exponential,expansional) and a Bayesian skyline plot (BSP, a non-parametric piecewise-constant model). The Bayes factor (BF) analysis showed that the relaxed clock fi tted the data signifi cantly better than the strict clock (2 lnBF=8 018 for relaxed clock). Under the relaxed clock the BF analysis showed that the exponential growth model was better than the other models (2 lnBF>7). Chains were conducted for at least 50×106generations, and sampled every 5 000 steps. Convergence was assessed on the basis of the eff ective sampling size after a 10% burn-in using Tracer software v. 1.5 (http://tree.bio.ed.ac.uk/software/ tracer/). Only parameter estimates with effective sampling size’s of>200 were accepted. Uncertainty in the estimates was indicated by 95% highest posterior density (95% HPD) intervals and the best fi tting models were selected by a BF (using marginal likelihoods)implemented in Beast[9]. The evolutionary distances among diff erent groups were calculated with MEGA6 software[10] using p-distance model. It was performed two diff erent type of evolutionary distance: the fi rst between clade Ⅰ and clade Ⅱ; the second between the subclades A1, A2 and B. The demographic history was also analyzed on the MVEV E gene sequences by performing the Bayesian skyline Plot to give an interpretation of the phylodynamic feature of the dataset.
3. Results
The phylogenetic noise of the dataset is shown in likelihood mapping (Figure 1). The percentage of dots falling in the central area of the triangles was 5.6%, and as the dataset showed no more than 30% of noise, it contained suffi cient phylogenetic signal.
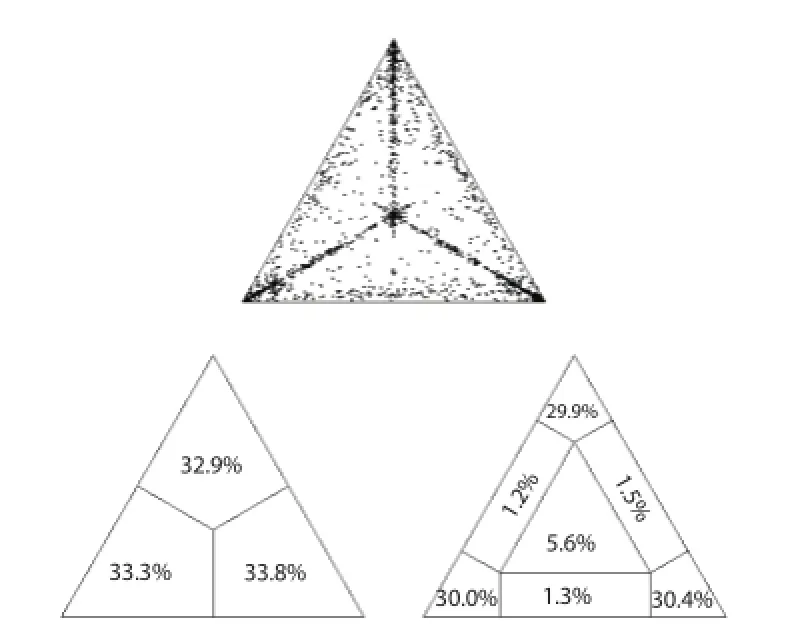
Figure 1. Likelihood mapping of MVEV E gene dataset.The dots inside the triangles represents the likelihood of the possible unrooted topologies for each quartet. Numbers indicate the percentage of dots in the centre of the triangle corresponding to phylogenetic noise (star-like trees).
The phylogenetic tree performed with PhyML 3.0, shows two clades highly signifi cant under a geographic selection (Figure 2). The clade Ⅰ including eight strains isolated from West Australia. The clade Ⅱ was characterized by at least four epidemic entries,three of which localized in NW Australia and one in Papua New Guinea. The genetic distance between the two clades (0.137 P=0.013) is a good suggestion for the hypothesis of two distinct epidemic entries. The Bayesian phylogenetic tree performed using a Bayesian MCMC approach is show in Figure 3.
The estimated mean evolutionary rate value of the MVEV E gene was 0.407×10-3substitution/site/year (95% HPD: 0.623×10-4-0.780×10-3). The root of the tree dated back to the year 1759. Two main clade were identifi ed, clade Ⅰ posterior probability (PP)=0.99 including the strains from Western Australia and dated back to the year 1948 (95% HPD: 1875-1995) and clade Ⅱ which included the strains from Papua New Guinea and the Australian coasts (PP=0.63)which dated to 1826 (95% HPD: 1643-1980).
Inside clade Ⅰ, subclade Ⅰ a (PP=0.81) dated to the year 1966(95% HPD: 1917-1997) and subclade Ⅰ b (PP=0.96) to the year 1980 (1978-1999). Inside clade Ⅱ, a major subclade (Ⅱ a), which dated back to the year 1939 (95% HPD: 1860-1989), can be found. Subclade Ⅱ a was characterized by different epidemic entries registered in diff erent time intervals. One epidemic entry was in 1980(95% HPD: 1938-2006) and was determined by two strains isolated from Papua New Guinea (labeled as PNG65298 and PNG69198)that grouped together, separately respect to the other sequences. Another entry was in 1952 (95% HPD: 1889-1990) and included the majority of the strains of subclade Ⅱ a.
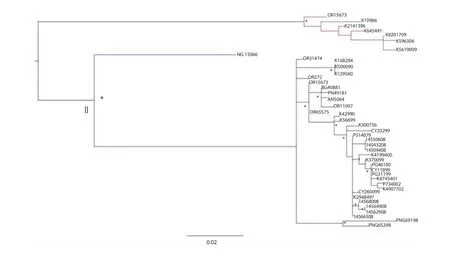
Figure 2. Maximum likelihood tree of the 46 MVEV E gene sequences.*: signifi cant statistical support for the clade subtending that branch (bootstrap support>70%). The scale bar indicates 2% of nucleotide divergence.
Specifically, inside the clade which dated back to 1969 (95% HPD: 1928-1990) a major clade (A) and a minor cluster (B) can be described. Inside clade A, two diff erent clusters (A1 and A2) were evident. Cluster A1 (PP=0.98) seems to have a spreading region around the West Cape York in the NE Australia, and is datableapproximately in the year 1992 (95% HPD: 1981-2000). Cluster A2 (PP=0.63) dated to 1996 (95% HPD: 1983-2005) included strains localized in the New South Wales region (SE Australia) and represents a separate and more recent entry of MVEV. Cluster B(PP=0.78) dated back to the year 1982 (95% HPD: 1954-1999) and included strains from NW Australian region. The genetic distances among these groups are shown in Table 1. The mean genetic distance showed that subclade A1 and A2 are closer than the other subclades(0.010 vs. 0.023 and 0.021).
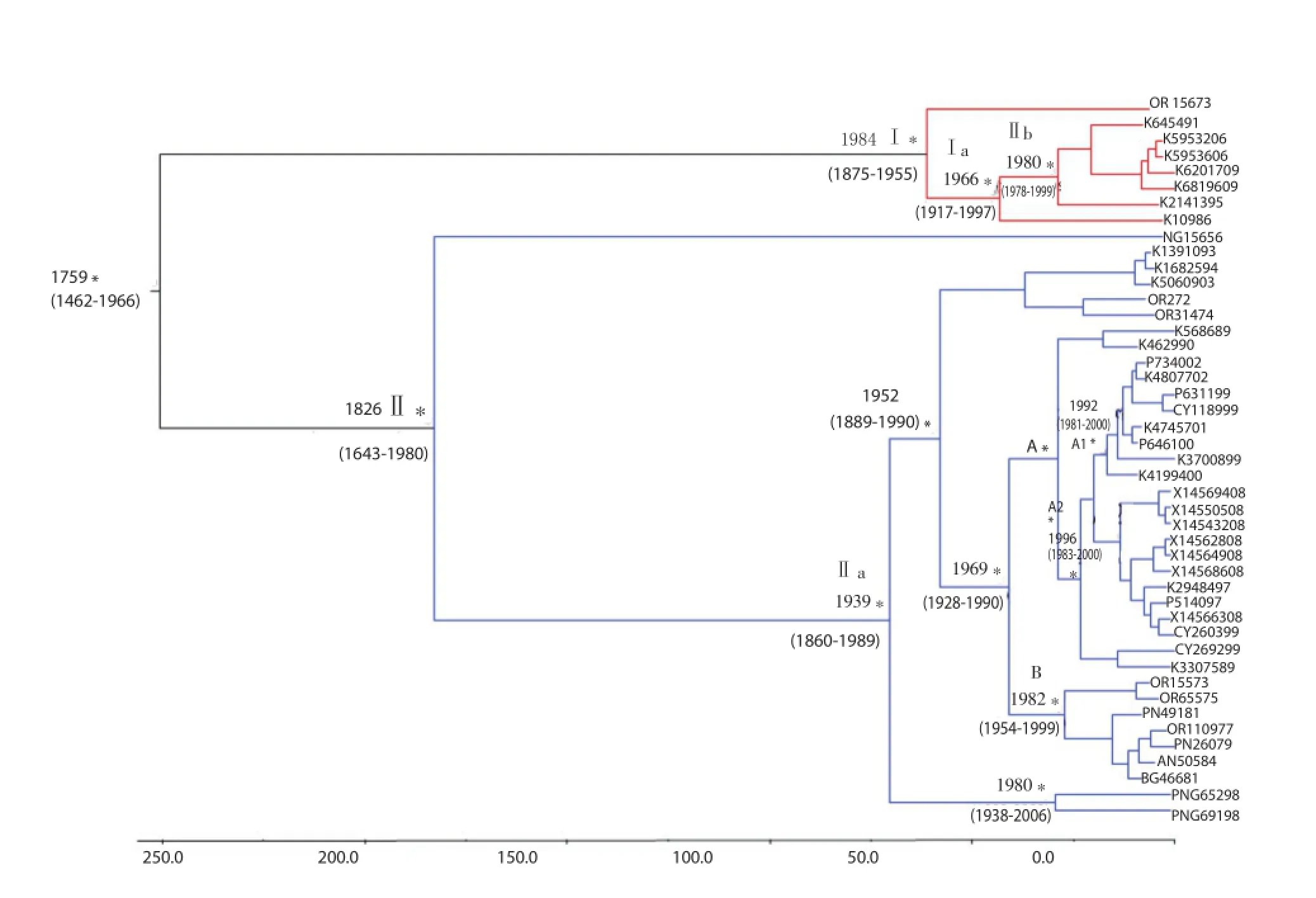
Figure 3. Bayesian phylogenetic tree of the MVEV E gene sequences.In diff erent colors: clade Ⅰ and Ⅱ; subclades A1, A2 and B. Time scale covers 250 years in the past beginning from year 2009 (last date of isolation strain in the dataset). The time of the most recent common ancestor, with the credibility interval based on 95% HPD, was reported in years. Scale years is reported at the bottom of the fi gure.

Table 1Estimates of evolutionary divergence over sequence pairs between subclades groups.
The BSP representing the estimates changes in eff ective population size through time is reported in Figure 4. The analysis of the BSP defi ned a relative constant population grow until the year 2000, when a strong reduction probably occurred due to a bottleneck.
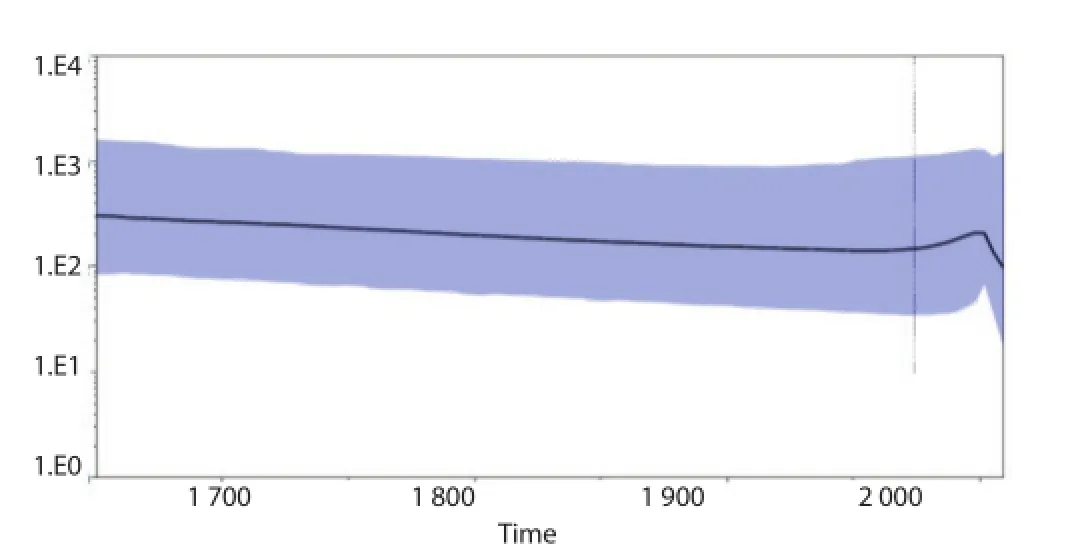
Figure 4. BSP of the MVEV E gene.The eff ective number of infections is reported on the Y-axis. The colored area corresponds to the credibility interval based on 95% HPD.
4. Discussion
This phylogenetic study provides diff erent suggestions about thediff usion and the biological history of MVEV in Australia and PNG. However previous studies highlighted the geographical distribution of this virus in Australia and PNG, the additionally evidences of the time-scale analysis defi ne a relative constant and well-localized presence of MVEV in these regions. The assumption that a prevalent distribution of the principal vector (Culex annilurostris) is defi ned by two strong conditions such as relative ecosystem and density population of humans in wetlands. These two condition suggested that this virus have maintained a constant fi tness and numerosity of its population for a lot of decades
As already described[5] the hypothesis of wide-spreading in Australia of MVEV is probably due to two factors: a first contribution of viraemic migratory water-birds[11,12] and a second contribution of wind-borne infected mosquitoes[13,14]. But since reduced density of waterbirds might explain the fewer MVEV case numbers in South-eastern Australia in 2011 compared to 1974, in spite of there being similar climatic conditions[15].
This is the fi rst study that estimated the mean evolutionary rate of MVEV E gene sequences and that reported the time-scaled phylogeny and phylodynamics of this virus. We estimated a mean evolutionary rate of MVEV E gene of 0.407×10-3substitution/ site/year (95% HPD: 0.623×10-4-0.780×10-3). Our time-scaled phylogeny reconstruction showed two main clades, labeled Ⅰ andⅡ, which dated back respectively to the year 1948 (95% HPD: 1875-1995) and 1826 (95% HPD: 1643-1980) indicating two distinct epidemic entries of this virus.
Moreover the phylodynamics analysis performed in the present study showed a relative costant population growth until the year 2000, followed by a reduction, probably due to climatic changes and decreasing circulation of the vectors. Climatic changes can gradually modify the ecosystem of Culex and waterbirds[16]. In the endemic regions of NW Australia the presence of MVEV population is only partially infl uenced by variation in rain-falling: in wetlands the intensity of rain-falling could have a detrimental eff ect to Culex larvae, decreasing the vector fitness; in arid grasslands an initial increasing of Aedes can leads to a more extensive viral diff usion.
The North to South diffusion of MVEV could probably increase from stronger tropical cyclone in northern WA activity. However, it is likely that some areas will have increases in arbovirus activity and human infection with predicted climate change, but risk of increased transmission will vary with locality, vector, host and human factors. In conclusion, our sophisticated phylogenetic methods have been useful in supporting the probable connection between climate changes and viral evolution also by the vector point of view. Moreover, considering climate changes and emerging importance of MVEV (as a JEV family member), it will be necessary to investigate and monitor this kind of outbreaks in order to prevent new viral epidemics inside and outside endemic areas.
Conflict of interest statement
We declare that we have no confl ict of interest.
[1] Marshall ID. Murray valley and Kunjin encephalitis. In: Monath TP,editor. The arboviruses: epidemiology and ecology. Boca Raton, FL: CRC Press; 1988, p. 151-189.
[2] Mackenzie JS, Broom AK. Australian X disease, Murray Valley encephalitis and the French connection. Vet Microbiol 1995; 46(1-3): 79-90.
[3] Bennett N. Murray Valley encephalitis: indeed a ‘mysterious disease’. Vic Infect Dis Bull 2008; 11: 94-107.
[4] Knox J, Cowan RU, Doyle JS, Ligtermoet MK, Archer JS, Burrow JN, et al. Murray Valley encephalitis: a review of clinical features, diagnosis and treatment. Med J Aust 2012; 196(5): 322-326.
[5] Johansen CA, Susai V, Hall RA, Mackenzie JS, Clark DC, May FJ, et al. Genetic and phenotypic diff erences between isolates of Murray Valley encephalitis virus in Western Australia, 1972-2003. Virus Genes 2007; 35: 147-154.
[6] Ciccozzi M, Ciccaglione AR, Lo Presti A, Yalcinkaya T, Taskan ZP,Equestre M, et al. Reconstruction of the evolutionary dynamics of the hepatitis C virus 1b epidemic in Turkey. Infect Genet Evol 2011; 11(5): 863-868.
[7] Zehender G, Ebranati E, Bernini F, Lo Presti A, Rezza G, Delogu M, et al. Phylogeography and epidemiological history of West Nile virus genotype 1a in Europe and the Mediterranean basin. Infect Genet Evol 2011; 11(3): 646-653.
[8] Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010;59(3): 307-321.
[9] Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007; 7: 214.
[10] Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 2013;30(12): 2725-2729.
[11] Porter JL, Kingsford RT. Aerial survey of wetland birds in Eastern Australia-October 2011 annual summary report. Sydney, NSW: Australian Wetlands, Rivers and Landscapes Centre. [Online] Available at: http://www.wetrivers.unsw.edu.au/wp-content/uploads/2012/06/2011-Eastern-Australia-Waterbird-Survey-Summary-Report.pdf 2011.
[12] Kingsford RT, Norman FI. Australian waterbirds-products of the continent’s ecology. EMU 2002; 102: 47-69.
[13] Kay BH, Farrow RA. Mosquito (Diptera: Culicidae) dispersal: implications for the epidemiology of Japanese and Murray Valley encephalitis viruses in Australia. J Med Entomol 2000; 37(6): 797-801.
[14] Ritchie SA, Rochester W. Wind-blown mosquitoes and introduction of Japanese encephalitis into Australia. Emerg Infect Dis 2001: 7(5): 900-903.
[15] Selvey LA, Dailey L, Lindsay M, Armstrong P, Tobin S, Koehler AP,et al. The changing epidemiology of Murray Valley encephalitis in Australia: the 2011 outbreak and a review of the literature. PLoS Negl Trop Dis 2014; 8(1): e2656.
[16] Russell RC, Currie BJ, Lindsay MD, Mackenzie JS, Ritchie SA, Whelan PI. Dengue and climate change in Australia: predictions for the future should incorporate knowledge from the past. Med J Aust 2009; 190(5): 265-268.
ent heading
10.1016/j.apjtm.2016.03.006
15 January 2016
Massimo Ciccozzi, Department of Infectious Parasitic and Immunomediated Diseases, Reference Centre on Phylogeny, Molecular Epidemiology and Microbial Evolution (FEMEM)/ Epidemiology Unit, National Institute of Health,Rome, Italy.
E-mail: ciccozzi@iss.it
Tel: +390649903187
in revised form 20 February 2016
ARTICLE INFO
Article history:
Phylogeny
Evolution
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Determination of ligand cluster and binding site within VP40 of Ebola virus: clue for drug development
- Clinacanthus nutans: a review of the medicinal uses, pharmacology and phytochemistry
- Current perspectives on dengue episode in Malaysia
- Etiological agents causing leptospirosis in Sri Lanka: A review
- Dengue outbreak in Swat and Mansehra, Pakistan 2013; an epidemiological and diagnostic perspective
- Bioactive extracts of red seaweeds Pterocladiella capillacea and Osmundaria obtusiloba (Floridophyceae: Rhodophyta) with antioxidant and bacterial agglutination potential