高效液相色谱-四极杆/线性离子阱质谱仪测定盐酸决奈达隆中两种基因毒性杂质的痕量残留
2016-11-08周长朋王东武张曼玉郑文凤
周长朋,王东武,张曼玉,许 洁,郑文凤
(迪沙药业集团国家认定企业技术中心,山东 威海 264205)
高效液相色谱-四极杆/线性离子阱质谱仪测定盐酸决奈达隆中两种基因毒性杂质的痕量残留
周长朋*,王东武,张曼玉,许洁,郑文凤
(迪沙药业集团国家认定企业技术中心,山东威海264205)
建立了高效液相色谱-四极杆/线性离子阱质谱仪(HPLC-QTRAP-MS/MS)测定盐酸决奈达隆药物中两种基因毒性杂质(杂质C和D)痕量残留的分析方法。盐酸决奈达隆药物以0.1%甲酸水溶液-乙腈(20∶80)溶解后,分别采用Agilent Extend-C18色谱柱(1.8 μm,2.1 mm×50 mm)或YMC Pack-CN色谱柱(5 μm,4.6 mm×250 mm)进行分离,以0.1%甲酸水和乙腈作为流动相分别进行梯度洗脱,电喷雾正离子(ESI+)扫描方式下选择离子监测(SRM)模式对样品进行检测。结果表明,杂质C和D在1.0~50 μg/L范围内线性关系良好,检出限(S/N=3)分别为0.20 μg/L和0.30 μg/L,定量下限(S/N=10)分别为0.80 μg/L和1.0 μg/L。该方法操作简单、灵敏度高、重现性好,可用于盐酸决奈达隆药物中两种基因毒性杂质痕量残留的测定。
高效液相色谱-四极杆/线性离子阱质谱仪;基因毒性杂质;盐酸决奈达隆;残留
盐酸决奈达隆(Dronedarone hydrochloride),化学名为N-[2-丁基-3-[4-[3-(二丁基氨基)丙氧基]苯甲酰基]-5苯并呋喃基]甲磺酰胺盐酸盐(合成路线见图1),是由法国赛诺菲-安万特公司开发的极化抑制剂,2009年7月首次在美国上市[1-2]。该物质是一种多通道阻滞剂,可同时抑制Na,K,Ca的离子通道,并具有β受体拮抗作用,可通过降低窦房结的自律性、减慢传导速度、延长动作电位时程和延长QT-QTC间期而产生抗心律失常作用,适用于心房颤动和心房扑动患者的心律控制、维持窦性心律和减慢室性心律[3]。该药与胺碘酮(Amiodarone)具有类似的电生理作用,但其亲脂性低,对甲状腺及甲状腺激素影响较小,无明显的心脏毒性,是新型的抗心律失常药物[4-5]。
基因毒性杂质在很低的浓度下即可诱导基因突变导致染色体断裂和重排,具有潜在的致癌毒性[6],现已引起广泛关注。国外已有多篇文献报道利用液相色谱-质谱联用仪检测药物中的基因毒性杂质[7-8],国内也有利用气相色谱-质谱联用仪[9-10]、液相色谱-质谱联用仪[11-12]等监控基因毒性杂质的方法报道。盐酸决奈达隆合成过程中产生的杂质C和D具有苯胺和硝基结构,依照欧盟基因毒性杂质指导原则[13],二者均属于潜在基因毒性杂质,需在产品中加以控制。基因毒性杂质限度为1.5 μg/d,1 d的服药量为800 mg,故杂质C和D的限度为1.875 μg/g,普通的液相色谱法测定很难达到限度[13-14],国内也未见利用液相色谱-质谱联用仪进行监控的报道。
四极杆-线性离子阱(QTRAP)质谱仪是混合型串联质谱,既保留了四极杆传统的定量功能,又可通过多级碎裂提供化合物的结构信息,具有高灵敏度和高特异性的特点,现已广泛应用于食品、药品、环境等领域[15-20]。本文基于高效液相色谱-四极杆/线性离子阱质谱(HPLC-QTRAP-MS/MS)技术,通过高分辨扫描后定性分析质谱图,并结合对照品定位确定了盐酸决奈达隆药物中的两种基因毒性杂质的结构,同时建立了测定盐酸决奈达隆药物中两种基因毒性杂质的分析方法。该方法简单、快速,回收率好、灵敏度高,填补了国内盐酸决奈达隆药物中基因毒性杂质检测研究的空白,有利于对盐酸决奈达隆药物合成过程中的质量参数进行有效监测。
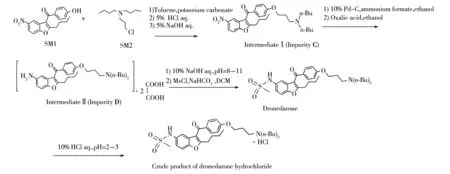
图1 盐酸决奈达隆的合成路线图Fig.1 Graphical synthetic route of dronedarone hydrochloride
1 实验部分
1.1仪器与试剂
Thermo LTQ-XL型高效液相色谱-四极杆/离子阱质谱仪:配有电喷雾离子源;超声波清洗机(昆山超声仪器公司);精密分析天平(OHAUS DV215CD);Milli-Q超纯水仪(美国Millipore公司);0.22 μm有机系滤膜(上海津腾公司)。
杂质C、杂质D对照品(迪沙药业集团有限公司,纯度≥99%);甲醇、乙腈(色谱级,德国Merck公司);甲酸(优级纯,天津大茂化学试剂厂);实验用水为Milli-Q超纯水(电阻率为18.2 MΩ·m);盐酸决奈达隆药品粗品和精品(迪沙药业集团有限公司)。
1.2溶液的制备
对照品溶液:取杂质C和D约10 mg,精密称定至10 mL容量瓶中,加少量乙腈超声溶解后,用乙腈定容至刻度,混匀,配制成1.0 g/L的标准储备液,-18 ℃避光保存。取适量标准储备液,用乙腈配制成10.0 mg/L的标准中间液,作为对照品溶液,4 ℃避光保存。
定性分析溶液:取盐酸决奈达隆粗品约10 mg,精密称定至10 mL容量瓶中,加少量乙腈超声溶解后再加入0.1%甲酸水溶液-乙腈(20∶80)定容。
供试品溶液:取盐酸决奈达隆精品约10 mg,精密称定至10 mL容量瓶中,加少量乙腈超声溶解后再加入0.1%甲酸水溶液-乙腈(20∶80)定容。以上溶液均需过0.22 μm滤膜后进行质谱分析。
1.3色谱条件
1.3.1定性检测色谱条件Agilent Extend-C18色谱柱(1.8 μm,2.1 mm×50 mm);流动相:A相为0.1%(体积分数)甲酸水溶液,B相为乙腈;流速:0.2 mL/min,等度洗脱(0~12 min,40%B);柱温35 ℃;进样体积为1.0 μL。
1.3.2杂质C定量检测色谱条件Agilent Extend-C18色谱柱(1.8 μm,2.1 mm×50 mm);流动相:A相为0.1%甲酸水溶液,B相为乙腈;流速:0.2 mL/min,梯度洗脱:0~1.0 min,15%B;1.0~5.0 min,15%~80% B;5.0~12.0 min,80%~15% B;柱温35 ℃;进样体积为10 μL。
1.3.3杂质D定量检测色谱条件YMC-Pack CN色谱柱(5 μm,4.6 mm×250 mm);流动相:A相为0.1%甲酸水溶液,B相为乙腈;流速:1.0 mL/min(柱后1∶4分流),等度洗脱(0~15 min,50%B);柱温35 ℃;进样体积为10 μL。
1.4质谱条件
1.4.1定性检测质谱条件离子源:电喷雾离子源(ESI);扫描方式:正离子扫描;检测方式:全扫描监测(SCAN);扫描范围:100~1 000;离子源温度:320 ℃;辅助气温度:300 ℃;鞘气(Sheath gas):35 L/min;辅助气(Aux gas):15 L/min;毛细管电压:3 500 V。

表1 杂质C和D的保留时间与质谱条件Table 1 Retention times and optimized spectrometric parameters of impurity C and impurity D
*quantification ion pairs
1.4.2定量检测质谱条件离子源:电喷雾离子源(ESI);扫描方式:正离子扫描;检测方式:选择离子监测(SRM);离子源温度:320 ℃;辅助气温度:300 ℃;鞘气:35 L/min;辅助气:15 L/min;毛细管电压:3 500 V;定量方式:外标法定量。具体质谱条件见表1。
2 结果与讨论
2.1定性分析与杂质结构确定
按照“1.3.1”和“1.4.1”条件制备定性分析溶液并进行检测,得到盐酸决奈达隆粗品的紫外色谱图和质谱离子流图(见图2),由图可见主峰后有两个杂质峰较为明显(峰2和峰3),并得到峰2和峰3的精确质荷比分别为m/z509.312 0和m/z479.413 0。通过仪器MassFrontier软件计算同位素位置和丰度比,得到两种离子的元素组成分别为C30H41N2O5+和C30H43N2O3+,即为C30H40N2O5和C30H42N2O3的[M+H]+信号。C30H40N2O5和C30H42N2O3的分子式分别与杂质C和D的分子式一致,将杂质C和D的对照品溶液按照上述条件检测后比对,保留时间和质荷比与峰2和峰3也完全一致,可判断峰2和峰3分别为杂质C和D,并在后期开展两种杂质的定量检测工作。

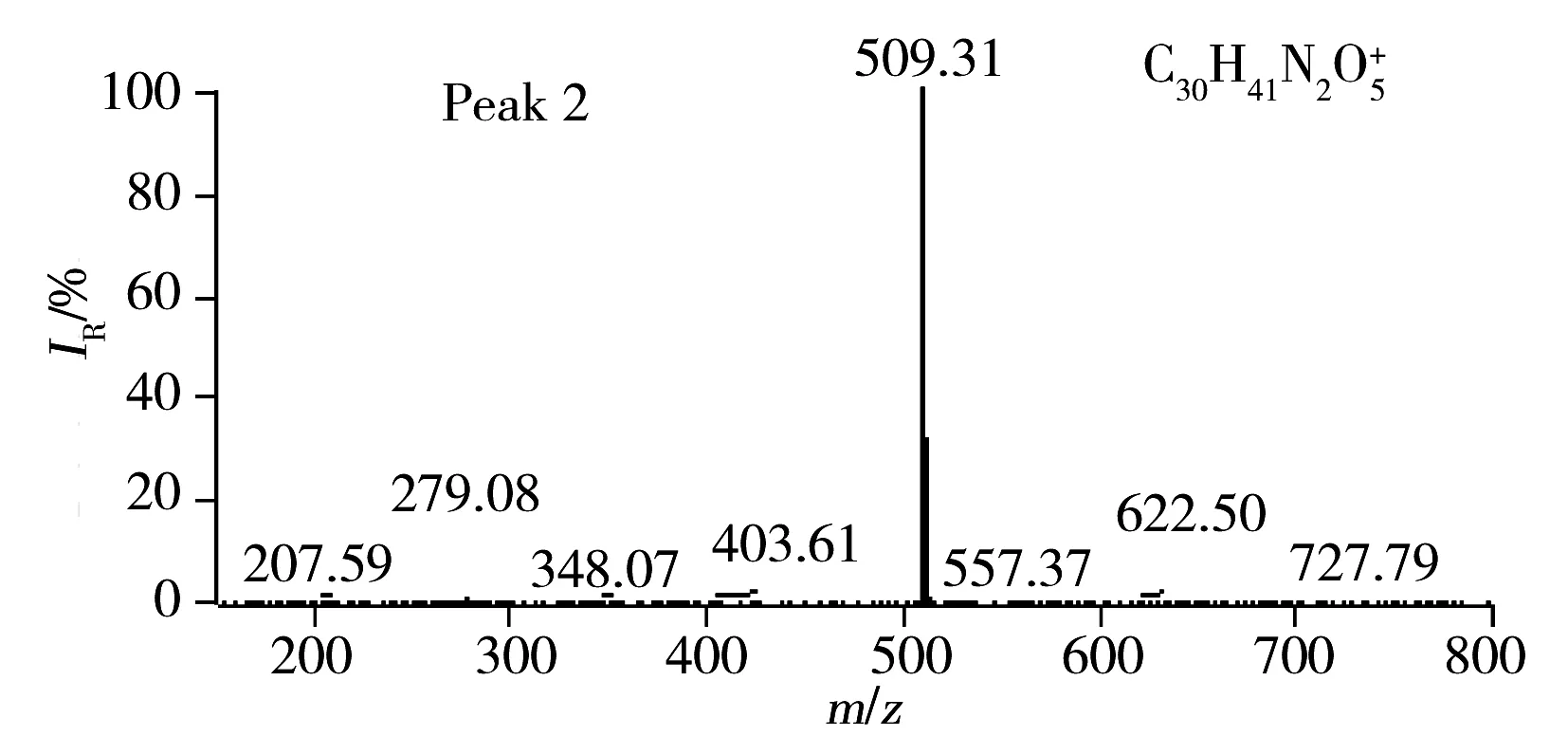
图2 盐酸决奈达隆粗品的色谱图及峰2、峰3的全扫描质谱图Fig.2 Chromatograms of dronedarone hydrochloride and scan mass spectra of peak 2 and peak 3
2.2定量检测色谱条件的选择
在相近的色谱条件下,考察了Agilent Extend-C18色谱柱(1.8 μm,2.1 mm×50 mm)、Thermo Hypersil-C18色谱柱(1.9 μm,2.1 mm×100 mm)和Agilent Eclipse XDB-C18色谱柱(3.5 μm,2.1 mm×100 mm)的分离情况。结果显示,在3种色谱柱上均能实现盐酸决奈达隆主峰和杂质C、杂质D的良好分离,且两种杂质的峰形均较好。但紫外图谱显示,盐酸决奈达隆主峰在后两种色谱柱上拖尾较为明显;进一步考察回收率时发现,在后两种色谱柱上杂质C的回收率明显降低。利用Agilent Extend-C18色谱柱能有效分离杂质C和主峰,且盐酸决奈达隆主峰无明显拖尾现象,杂质C峰形好、回收率高,故本文采用Agilent Extend-C18色谱柱检测盐酸决奈达隆中杂质C的残留。
通过回收率实验发现杂质D在上述3种色谱柱上分离检测后回收率均很低,调节梯度洗脱浓度使主峰和杂质D分离度增大后回收率也无明显改善,可能的原因是盐酸决奈达隆高浓度主峰对杂质D有强烈的离子抑制效应。尝试利用氰基柱(YMC-Pack CN色谱柱)对二者进行分离,杂质D在盐酸决奈达隆主峰前2 min出峰,峰形较好;回收率实验进一步表明,杂质D在主峰前出峰时的回收率良好,基本在80%以上,故本文采用氰基柱(YMC-Pack CN色谱柱)检测盐酸决奈达隆中杂质D的残留。
2.3质谱条件的选择
采用电喷雾离子源,在正离子模式下,对1.0 mg/L杂质C和杂质D标准溶液进行母离子全扫描,确定其分子离子[M+H]+峰(m/z509.3和479.4);对分子离子峰进行子离子扫描,得到二级质谱图,筛选响应值较高、基线噪音低的两个离子作为定性离子对。采用SRM模式采集数据,驻留时间设定为100 ms,优化各离子的碰撞能,得到最佳质谱条件如表1所示。
2.4线性范围、检出限及定量下限
以0.1%甲酸水溶液-乙腈(20∶80)配制0,1.0,5.0,10,20,50 μg/L的杂质C和杂质D标准系列溶液分别进行测定,以定量离子峰面积对质量浓度进行线性回归。结果表明,杂质C和杂质D在1.0~50 μg/L范围内线性关系良好(r>0.998);以S/N=3计算检出限分别为0.20 μg/L和0.30 μg/L;以S/N=10计算定量下限分别为0.80 μg/L和1.0 μg/L。方法的灵敏度完全满足依照欧盟基因毒性杂质指导原则计算的1.875 μg/L的杂质限度。
2.5回收率实验
采用不含杂质C和杂质D的盐酸决奈达隆精品为空白基质,添加80%限度、100%限度、120%限度浓度的杂质C和杂质D分别做回收率实验,平均回收率分别为87.8%~96.8%和82.0%~88.0%,相对标准偏差(RSD)分别为1.5%~5.0%和2.0%~2.7%(见表2)。
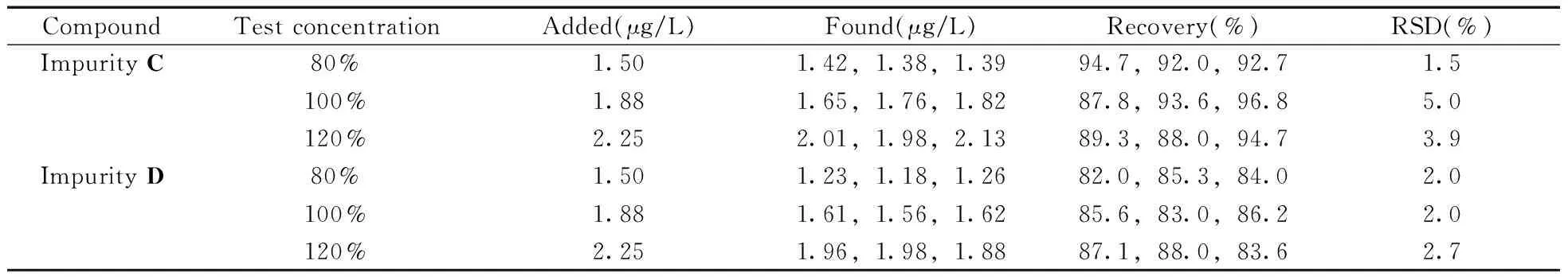
表2 盐酸决奈达隆中添加杂质C和杂质D的回收率与相对标准偏差Table 2 Recoveries ans RSDs of impurity C and impurity D in dronedarone hydrochloride
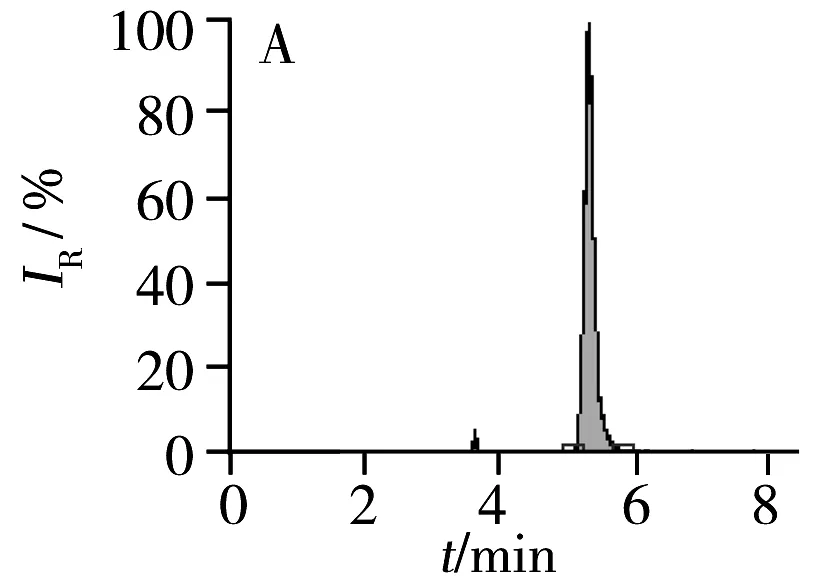
图3盐酸决奈达隆中含杂质C(A)和杂质D(B)的阳性样品总离子流图(TIC)
Fig.3Total ion chromatograms(TIC) of positive samples with impurity C(A) and impurity D(B) in dronedarone hydrochloride
2.6实际应用
将所建方法应用于合成的盐酸决奈达隆药物小试3批、中试3批及工艺验证3批共9份样品的检测,检出2批含有杂质C残留,含量分别为3.42 mg/kg和1.93 mg/kg;6批含有杂质D残留,含量为3.82~148.2 mg/kg,其他样品均未检出。阳性样品的色谱图见图3。
3 结 论
本文建立了高效液相色谱-四极杆/线性离子阱质谱仪(HPLC-QTARAP-MS/MS)测定盐酸决奈达隆药物中两种基因毒性杂质(杂质C和杂质D)痕量残留的分析方法。利用线性离子阱质谱的高分辨扫描功能联合对照品定性成功确定了两种杂质结构,并建立了定量检测方法。该方法灵敏度高、分离度好,回收率、精密度及定量下限均能满足基因毒性杂质限度的要求,是检测盐酸决奈达隆药品中两种基因毒性杂质痕量残留的有效方法。将所建方法应用于合成的盐酸决奈达隆药物样品的检测,共检出2批样品含有杂质C,6批样品含有杂质D,有效指导了盐酸决奈达隆合成反应的进行。
[1]He X Q,Wu T Z,Zhang F L,Xie M H.Chin.J.Pharm.(何晓清,吴泰志,张福利,谢美华.中国医药工业杂志),2010,41(2):148-151.
[2]Wang G,Zhang F L,Wang J J,Li J Q.Chin.J.Pharm.(王冠,张飞龙,王佳静,李建其.中国医药工业杂志),2011,42(12):881-883.
[3]Xu L F,Jiang M J,Liu J,Wang X J,Wang Y,Chen Y,Zhou Y P.J.LiaoningUniv.:Nat.Sci.Ed.(徐利锋,姜明俊,刘举,王雪娇,王洋,陈烨,周云鹏.辽宁大学学报:自然科学版),2012,39(2):103-109.
[4]Liu J,Qin Y W.J.Pharm.CareRes.(刘璟,秦永文.药学服务与研究),2008,8(6):417-420.
[5]Li F,Tian S H,Song X F,Li W Y,Cheng H Y,Lei L Z,Zhu Q F,Jin C H,Li S H.Chem.J.Chin.Univ.(李峰,田拴红,宋晓峰,李文远,程花英,雷灵芝,朱勤丰,金春华,李少华.高等学校化学学报),2013,34(4):858-862.
[6]Ma L,Ma Y N,Chen Z,Jiang Y.Chin.J.NewDrugs(马磊,马玉楠,陈震,蒋煜.中国新药杂志),2014,23(18):2106-2111.
[7]Vijaya Bhaskar Reddy A,Venugopal N,Madhavi G,Gangadhara Reddy K,Madhavi V.J.Pharm.Biomed.Anal.,2013,84:84-89.
[8]Venugopal N,Vijaya Bhaskar Reddy A,Gangadhar Reddy K,Madhavi V,Madhavi G.J.Pharm.Biomed.Anal.,2012,70:592-597.
[9]Ding Y M,Wu J,Lu C X,Cui P,Wang D C.Chin.J.Pharm.(丁逸梅,吴娟,陆春晓,崔萍,王德才.中国医药工业杂志),2014,45(11):1069-1071.
[10]Liu L Y,Zhang Q R,Sun L F,Wang M M,Guo W M.Chin.J.Pharm.(刘立云,张倩如,孙连福,王曼曼,郭文敏.中国药师),2014,17(8):1274-1276.
[11]Zhang L,Huang L N,Li Q R,Yang W H.Chin.J.Pharm.Anal.(张莉,黄丽娜,黎其荣,杨伟涵.药物分析杂志),2015,35(2):317-319.
[12]Liu J H,Zhong Y N,Xiong Y,He W T.Chin.J.Pharm.Anal.(刘俊华,钟雅妮,熊渊,赫涡涛.药物分析杂志),2013,33(7):1168-1170.
[13]European Medicines Agency.Guideline on the Limits of Genotoxic Impurities[EB/OL].http://www.ema.eu- ropa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002903.pdf,2006-06-28/2013-12-13.
[14]Jin P F,Liang X L,Li Q,Zou D,Ma J,Hu X.Chin.J.Pharm.Anal.(金鹏飞,梁晓丽,李琼,邹定,马捷,胡欣.药物分析杂志),2013,33(3):450-453.
[15]Luo Y Y,Liu J X,Liu X H,Lan C W,Hou Y,Ma Y,Xu L.J.Instrum.Anal.(罗益远,刘娟秀,刘训红,兰才武,侯娅,马阳,徐力.分析测试学报),2015,34(5):519-524.
[16]Zhou Y,Zhao Y G,Zhang B B,Zhang Y,Chen G S.Chin.J.Anal.Chem.(周岩,赵永刚,张蓓蓓,章勇,陈国松.分析化学),2014,42(3):367-374.
[17]Ruan X L,Zhang A H,Wu C,Rong W F,Huang S L.J.Instrum.Anal.(阮小林,张爱华,吴川,戎伟丰,黄淑莲.分析测试学报),2011,30(1):72-75.
[18]Yang J J,Wu S Q,Tong L,Song S L,Tian Q.J.Instrum.Anal.(杨佳佳,吴淑琪,佟玲,宋淑玲,田芹.分析测试学报),2011,30(4):374-380.
[19]Liu J,Zhang M,Wan Y,Wu H M,Hou Y N,Zheng L M,Yin Y X.Chin.J.Anal.Chem.(刘佳,张朦,万有,吴红梅,侯艳宁,郑乐民,尹玉新.分析化学),2014,43(8):1118-1124.
[20]Zhang H W,Jian H M,Lin L M,Chen L Z,Liang C Z,Yuan T,Tang Z X,Cai X,Qin L Y.J.Instrum.Anal.(张鸿伟,简慧敏,林黎明,陈亮珍,梁成珠,袁涛,汤志旭,蔡雪,秦良勇.分析测试学报),2012,31(7):763-770.
Determination of Two Genotoxic Impurities Residues in Dronedarone Hydrochloride by HPLC-QTRAP-MS/MS
ZHOU Chang-peng*,WANG Dong-wu,ZHANG Man-yu,XU Jie,ZHENG Wen-feng
(National Enterprise Technology Center of Disha Pharmaceutical Group,Weihai264205,China)
A high performance liquid chromatography-quadrupole ion mass spectrometric(HPLC-QTRAP-MS/MS) method was developed for the determination of two potential genotoxic impurities(impurity C and impurity D) residues in dronedarone hydrochloride.After extracted with 0.1% formic acid-acetonitrile(20∶80),the dronedarone hydrochloride samples were separated on an Agilent Extend-C18column(1.8 μm,2.1 mm×50 mm) or a YMC Pack-CN column(5 μm,4.6 mm× 250 mm) with two mobile phases of 0.1% formic acid and acetonitrile.The detection of analytes was performed in the positive mode and selective reaction monitoring(SRM) mode.As a result,the calibration curves of impurity C and impurity D showed good linearity in the range of 1.0-50 μg/L.The limits of detection(S/N=3) were 0.20 μg/L and 0.30 μg/L,respectively.The limits of quantitation(S/N=10)were 0.80 μg/L and 1.0 μg/L,respectively.The method has the advantages of simple operation,high sensitivity,good reproducibility,and could be used for the detection of two genotoxic impurities residues in dronedarone hydrochloride.
high performance liquid chromatography-quadrupole ion mass spectrometry(HPLC-QTRAP-MS/MS);potential genotoxic impuritie;dronedarone hydrochloride;residues
2016-02-02;
2016-02-19
周长朋,工程师,研究方向:理化检测及药物分析,Tel:0631-3857373,E-mail:20019568@qq.com
10.3969/j.issn.1004-4957.2016.09.006
O657.63;TQ460.72
A
1004-4957(2016)09-1111-05
