浓香型白酒窖泥变质前后真菌群落差异分析
2016-11-04于春涛刘超陈瑞玲朱凤林刘振江
于春涛,刘超,陈瑞玲,朱凤林,刘振江
(1.沧州医学高等专科学校,河北沧州061001;2.河北省宁晋县泥坑酒业有限责任公司,河北石家庄054000)
浓香型白酒窖泥变质前后真菌群落差异分析
于春涛1,刘超1,陈瑞玲1,朱凤林1,刘振江2
(1.沧州医学高等专科学校,河北沧州061001;2.河北省宁晋县泥坑酒业有限责任公司,河北石家庄054000)
窖泥变质是北方酒厂面临的重大难题,通过直接提取窖泥真菌总DNA,利用高通量测序的方法扩增18S rDNA并构建基因文库,分析窖泥变质前后真菌群落组成。结果表明,窖泥变质前基因序列数为9737条,归为2个门,优势真菌主要为:小囊菌属(74.21%)、青霉菌属(10.03%)、假裸囊菌属(9.99%)等;窖泥变质后基因序列数达到了78322条,归为19个门,优势真菌主要为:青霉菌属(76.56%)、假裸囊菌属(6.73%)、纤孔菌属(5.96%)、丝盖伞属(2.93%)等。窖泥变质前后真菌的种类和丰度均有显著差异,为全面了解窖泥变质提供了理论依据。
窖泥;变质;真菌群落;差异分析;高通量测序
浓香型白酒的生产以窖池中的窖泥为基础,窖池中庞大的微生物群落在发酵过程进行着复杂的物质代谢和能量代谢,从而促进了窖泥老熟和酒质的提高[1]。法国人A.Calmette较早开展酒类微生物的研究,建立了可以利用淀粉发酵生产酒精的“阿米露法”[2]。我国浓香型白酒厂分布广、数量多,关于菌群结构及其变化规律的研究成果更为丰富。廖建民等[3]对多个国家名优酒厂不同季节、不同贮存期的样本进行了研究,获得真菌94株,其中霉菌69株、酵母菌25株。窖泥中功能真菌具有产酒精、产酶及产香作用[4]。为了揭示“窖泥产香”的奥秘,我国学者对窖泥功能微生物进行了大量的分离、纯化和鉴定工作,目前从窖泥中分离的微生物以细菌为主,对真菌研究很少。然而如果对窖泥管理不善,窖泥中微生态平衡被破坏,微生物代谢出现异常,致使窖泥出现板结变硬、腐败气味等变质现象[5-7],导致酒质下降,没有窖香,不具有浓香型白酒的风格,会给酒厂带来很大的经济损失。因此研究窖泥真菌群落的动态变化,检测出窖泥变质前后主要优势真菌,对人工窖泥培养和提高浓香型白酒质量尤为重要。
由于自然界中只有0.1%~10%的微生物可以直接培养,传统分离培养方法难以全面准确地反映窖泥生态系统微生物的真实情况[8-9]。近年来随着微生物生态学的发展,分子生物学技术越来越多的应用于窖泥微生物群落结构和动态变化规律的研究,不少学者研究分析窖泥的群落组成,如罗惠波[10]、黄治国[11]等通过PCR-SSCP结合聚类分析技术比较泸州老窖不同的窖龄的窖泥中细菌和古菌的群落差异。邓依等[12]通过细菌rDNA ITSAFLP结合聚类分析技术比较了多粮发酵新老窖池间的细菌群落差异。但是对窖泥变质前后具体的真菌群落结构的报道比较少。
Illumina Solexa合成测序是近几年发展起来的一项全新的高通量DNA测序技术,具有高准确性、高通量、高灵敏度和低运行成本等突出优势,可以同时完成传统基因组学研究(测序和注释)以及功能基因组学(基因表达及调控,基因功能,蛋白/核酸相互作用)研究,已广泛应用于探索土壤、海洋和湿地,以及食品发酵和废水处理反应器等微生物区系研究,并取得了丰硕的研究成果[13-14]。目前,利用高通量测序技术对窖泥中真菌群落结构的研究还未见文献报道。本研究利用该方法对窖泥变质前后真菌群落结构进行了分析,为全面、系统地揭示窖泥变质前后微生物群落结构提供理论依据。
1 材料与方法
1.1材料
样品采集:窖泥样品取自河北某知名酒厂酿酒车间优质窖泥(FNK1)和变质窖泥(PNK1)。取样方法是FNK1从优质窖池的窖壁和窖底各取3点样共100 g,PNK1从变质窖池的窖壁和窖底各取3点样共100 g,分装后密封冷冻保存。
主要试剂:E.Z.N.A.Soil DNA Kit(OMEGA)、Qubit2.0 DNA检测试剂盒(Life)、Taq DNA Polymerase(Thermo)、SanPrep柱式DNA胶回收试剂盒(上海生工)。
1.2实验方法
1.2.1窖泥预处理及总DNA提取[15]
称取0.2 g样品放入含有0.5 g磁珠的5 mL离心管中,加入1 mL SLX-mLus Buffer,漩涡2 min。经OMEGA土壤DNA提取试剂盒,反复冻融、离心,收集上清液。粗提的窖泥DNA通过PCR产物纯化试剂盒纯化,纯化后的窖泥DNAPCR扩增效果良好。
1.2.2PCR扩增及对产物进行琼脂糖凝胶电泳
利用Qubit2.0 DNA检测试剂盒对基因组DNA精确定量,以确定PCR反应应加入的DNA量。PCR所用的已经融合了Miseq测序平台的NS1-Fung通用引物:NS1引物:CCTACACGACGCTCTTCCGATCTN(barcode)GTAGTCATATGCTTGTCTC,Fung引物:GACTGGAGTTCCTTGGCACCCGAGAATTCCAATTCCCCGTTACCCGTTG。PCR体系按照如下进行:10×PCR buffer 5 μL,dNTP(10 mM each)0.5 μL,Genomic DNA 10 ng,Bar-PCR primer F(50 uM)0.5 μL,Primer R(50 uM)0.5 μL,Plantium Taq(5 U/μL)0.5 μL,H20 add to 50 μL。配制好的PCR体系按照如下反应条件进行扩增:94℃变性3 min,接下来进行5个循环,分别是94℃变性30 s,45℃退火20 s,65℃延伸30 s;再接着25个循环,分别是94℃变性20 s,55℃退火20 s,72℃延伸30 s,72℃延伸5 min,然后进行第2轮扩增,引入lllumina桥式PCR兼容引物。PCR结束后,产物进行琼脂糖凝胶电泳,对DNA进行回收。
1.2.318S rDNA扩增片段的测序、质控和聚类[16-18]
采用lllumina Miseq测序平台对扩增片段进行双端测序,测得的基因序列经质量控制软件Prinsep处理,去除barcode、两端primer以及部分低质量基因序列。
对测得的基因序列采用uclust软件进行OTU聚类,通常域值的序列相似性定位0.97,操作分类单元被认为可能属于属。
1.2.4对处理后序列进行物种分类,比较窖泥变质前后真菌结构差异
物种分类采用的软件为RDP classifier,基于OTU聚类的结果,获取每一个OTU聚类的代表序列,分别是长度最长序列(length)、丰度最大序列(abundance)和所有序列(ALL)形成3份结果,并对各类RDP分析。本文所有的展示,均使用OTU_ALL中的数据,genus水平进行展示,采用柱形图及表格的形式对比窖泥变质前后群落结构差异。
1.2.5MEGAN分析
使用MEGAN软件,通过交互式搜索NCBI中的分类数据库信息,以树状图形式表现物质丰度情况与菌落结构,反应窖泥变质前后真菌的组成情况。
2 结果与分析
2.1窖泥变质前后真菌18SrDNANS1-Fung电泳图谱(图1)
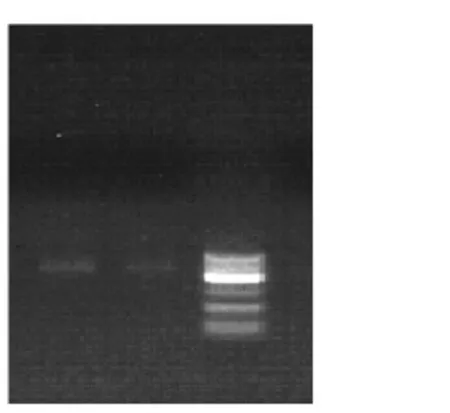
图1 真菌18S rDNANS1-Fung电泳图谱
由图1所示,3条泳道对比得知,泳道1和泳道2有较清晰的条带,DNA提取时一般都通过试剂盒过柱提取,这种柱子有固定孔径,从而决定了基因组片段的大小。泳道1比泳道2亮,说明窖泥变质后真菌数量和丰度均有所提高。基因序列长度大部分分布在400~600 bp之间,基本满足分析需要。
2.2窖泥变质前后18S rDNA多样性分析
2.2.118S rDNA处理后序列数
扩增后的PCR产物去除嵌合体和靶区域序列,得到的基因序列数见表1。
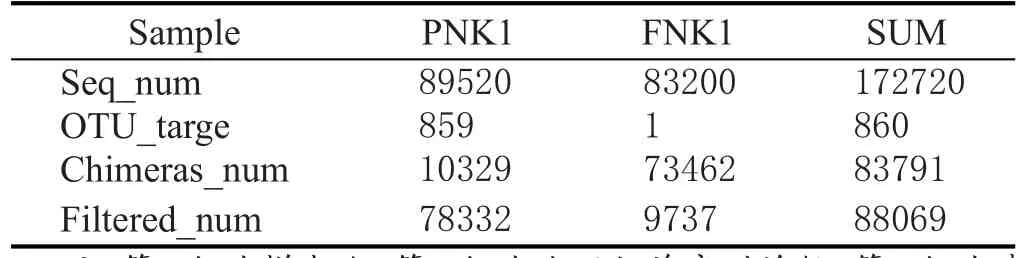
表1 处理后基因序列统计表
综合表1处理后的基因序列数表明,窖泥变质后的序列数显著高于变质前,说明窖泥变质后真菌的种类和丰度有明显的增多。
2.2.2OTU聚类分析
将多条序列按其序列间的距离对它们进行聚类后,根据序列之间的相似性作为域值分成操作分类单元(OTU),通过OTU数目变化与uclust参数similarity值之间的数据对比,从中选择最佳的similarity值为0.97,操作分类单元被认为属于属。采用OTU VENN软件分析样本中PNK1有861个OTUs,FNK1有229个OTUs,两者同时有228个OTUs(图2)。PNK1增加了633个OTUs,表明它们可能是在酿酒过程中,由于操作不当而新进的菌种。这些真菌群落可能与窖泥变质有关。
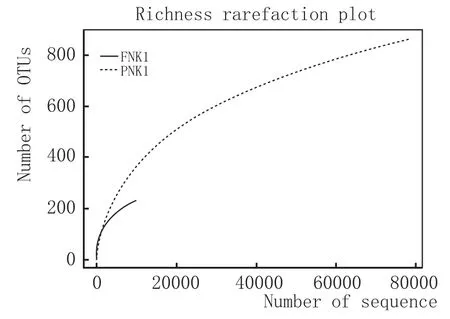
图2 FNK1和PNK1 OTUS对比
2.3真菌群落结构分析
对处理后序列进行物种分类,采用RDP classifier软件对每条序列在genus水平上计算其分配到此rank中的概率值,一般概率值大于0.8,即RDP分类域值[19-21]。根据分类学分析结果,可以得知样品在属分类水平的数据(表2、表3)。
结合表2、表3所示,窖泥变质前后真菌群落结构、丰度差异较大,选择优势菌群进行比较。窖泥变质前绝对优势菌属为小囊菌属(Microascus),窖泥变质后青霉菌属(Penicillium)成为绝对优势菌属。Thomton[22]在前人研究的基础上,得出结论:在未经扰动的土地上,小部分优势菌种是特定有利环境的结果,这个结论被普遍接受。每个研究的小生境内环境是相似的,其中窖泥真菌区系也相似,大量事实证实了窖泥真菌组成与生境类型有明显的对应性。影响生境选择的可能因子很复杂,Widden[23]通过多变量分析的方法(多元回归,典型相关分析,因子分析,分类排序等),证实了真菌种类的出现与生物及非生物环境之间有复杂的多重关系。非生物因子如Ca2+、温度、湿度、K+、窖泥的形成过程等,可能是少数或几个主要真菌种出现的首要调节者。但有更多的证据证实生物因子有可能是主要的调节者。Gochenaur[24]研究后得出结论:种间竞争在调节种的丰富性方面是最主要的因子。窖泥中的真菌群落在经历一个长期驯化、变化过程中,由于窖泥化学特性和窖泥生物的改变,再加上外来生物因素的影响,窖泥中的真菌群落种类组成发生改变,新的真菌群落更能适应新的环境。窖泥变质后青霉菌属为主要调节者,通过种间相互竞争得到了如表3所示的真菌群落结构。

表2 FNK1真菌种类及丰度值
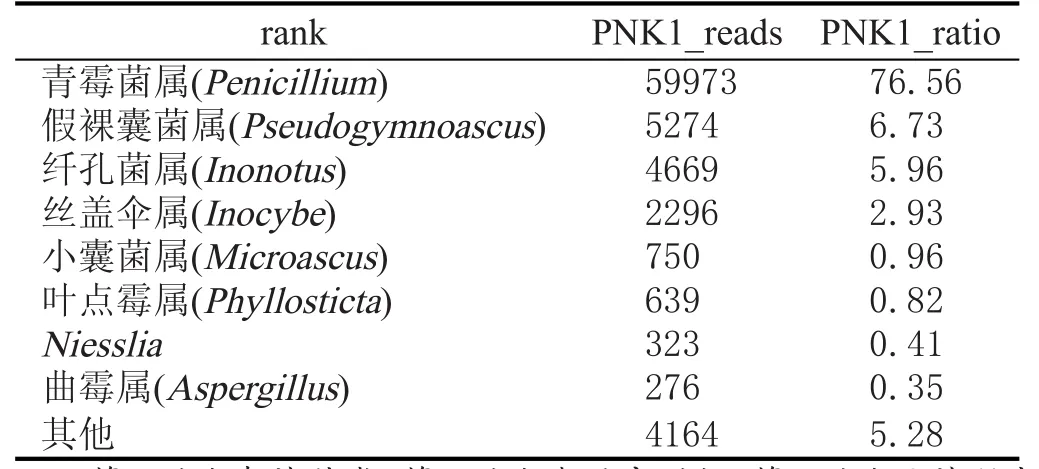
表3 PNK1真菌种类及丰度值
2.4窖泥变质前后真菌分类树
使用MEGAN软件,通过交互式搜集NCBI中的分类数据库信息,以树状图形式在门水平表现窖泥变质前后真菌群落结构(图3、图4)。
图3 FNK1门水平MEGANF丰度图
如图3所示,窖泥变质前真菌群落结构只有子囊菌门(99.99%)和担子菌门(0.01%),来源于原始窖泥和人工驯化。窖泥中的真菌除了能起到酿酒产香的作用外,在微生态系统中也发挥着多种多样的功能,如降解纤维素、半纤维素、木质素、胶质、还原氮、溶解磷、螯合金属离子、产生青霉素等。然而在Dobranic和Zak[25]1999年提出FungiLog理论框架和方法学之前,土壤真菌的功能多样性研究仅仅停留在个别真菌种类和类群功能的研究方面,缺乏系统性和理论性。由于存在分离和检测技术的限制,迄今为止仅有少数窖泥真菌被报道,窖泥真菌群落之间的理化特性也就不得而知。根据Gochenaur[24]所提出的理论,土壤中优势菌属是微生态的主要调节者,因此在窖泥变质前建立了以子囊菌门中的小囊菌属为主要调节者的真菌群落结构。如果小囊菌属发生变化,则整个真菌群落结构将发生变化。
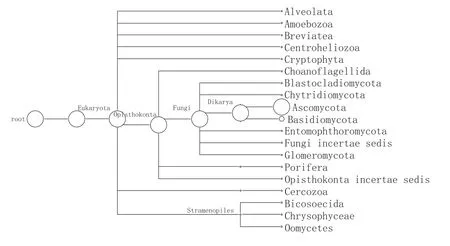
图4 PNK1门水平MEGANF丰度图
由图4所示,窖泥变质后微生态发生了很大变化。共鉴定出10个真菌门、3个藻类门和其他后鞭毛生物界的6个门。其中子囊菌门(89.36%)和担子菌门(9.62%)为主要优势真菌。窖泥真菌群落组成与生长环境有明显的对应性,随着酿造周期的增加,窖泥中真菌的生长环境(如温度、湿度、营养等)发生变化,从而对应的真菌群落也将发生变化。20世纪60年代以前,少有关于土壤真菌与环境演替关系的报道,较早的如Mallik[26]研究了俄克拉荷马代表森林植被不同演替阶段区域的土壤真菌。随着演替的进行,窖泥湿度、有机碳、Ca2+、K+等因子都逐渐升高,真菌的种类也增多。
3 讨论
随着真菌研究的不断深入,传统纯培养技术和显微镜观察方法已经不能适应研究的需要,Hawksworth[27]研究表明,自然界中约有150万真菌种类,但仅有0.7%能被分离和培养。随着分子生态学方法的建立和不断改进,采用Lllumina Miseq测序平台的高通量DNA测序技术,对窖泥变质前后真菌群落组成、结构、动态方面有了更深入的了解,为研究窖泥功能真菌及窖泥变质的真菌影响提供有利支持。此种方法还能够分析真菌群落和环境因子之间的关系,为将来通过检测真菌群落结构推测环境因子打下基础。
通过18S rDNA序列分析表明,窖泥变质前后18S rDNA扩增序列为9137条和78332条,聚类结果为229个OTUs和861个OTUs,说明真菌的种类和丰度在窖泥变质后均有较大的提高。窖泥的物理环境条件如土壤质地、pH值、C/N值、光照、水分、温度等因素对真菌群落产生一定的影响,它们或是单个因子或是多个因子共同起作用影响着窖泥真菌的群落组成。有研究报道,土壤真菌数量随水分的增加而增加[28],巨天珍等[29]研究认为,影响土壤真菌数量和多样性的最主要因素是pH值,其次是土壤中的有机质和水分含量。Paul等[30]主张对土壤真菌群落变化有关的是总C/N值,次要的因素是土壤pH值。Sabine[31]认为,土壤真菌群落组成更多的是依赖于所在生境的有效性,因为有效的生境有利于分散的孢子和繁殖体在土地利用方式改变后容易定植。吴敏娜等[32]研究表明,细菌的群落结构也会影响真菌的生长,随着土壤细菌群落结构越来越偏离自然清洁土壤,其抑制真菌作用逐渐降低,表明细菌群落结构与土壤抑制真菌作用之间的相关性更为密切。
采用RDP classifier软件对18S rDNA序列分析得知,窖泥变质前优势菌属为小囊菌属(74.21%),窖泥变质后优势菌属为青霉属(76.56%)。青霉属[33]成员广泛存在于自然界,有多种多样的代谢产物,能产生有机酸、青霉素等,青霉菌也可以使食品、水果、饲料、种子、烟草、药材发生酶腐变质,腐蚀工业器材、仪器设备和原料是工业和实验室污染源之一。自然界中已发现的青霉绝大多数以无性繁殖的方式繁衍后代,分生孢子萌发菌丝体,气生菌丝产生分生孢子梗,然后生出许多分生孢子,分生孢子在适宜环境中又萌发为菌丝体,以此循环反复,导致变质后窖泥中青霉属成为绝对优势菌属,青霉属是否在窖泥变质过程中起到直接或间接作用,有待于认识窖泥微生物全貌再作综合分析。
真菌作为窖泥重要的生物类群,对白酒酿造及窖泥微生态都起着重要作用。由于真菌个体小、数量多,其多变的类型和强大的适应环境能力为我们了解窖泥真菌全貌带来障碍。通过高通量DNA测序技术可以快速直观地得到窖泥真菌的多样性并且获得大量未知种类,显示出分子生物技术的优势,然而作为一种实验手段,此种技术并不是万能的,因为影响PCR的因素限制了其应用,因此应保留传统培养方式。研究窖泥真菌群落结构及其生理生化特性、形态结构,必须将传统培养方法和现代分子生物学方法结合起来。
[1]张文学,乔宗伟,胡承,等.浓香型白酒糟醅中真菌菌群的多样性分析[J].四川道大学学报(工程科学版),2006,38(5):97-101.
[2]陈宪仪.生物与中国酿造文化[J].中国酿造,2001(3):33-35.
[3]廖健民,任道琼,唐玉明,等.浓香型曲药中酵母菌的初步分类和选育[J].酿酒,2000,137(2):48-48.
[4]刘雯雯,孙剑秋,平文祥,等.中国白酒窖池真菌的研究进展[J].中国酿造,2012,31(6):1-5.
[5]魏景超.真菌鉴定手册[M].上海:上海科学基础出版社,1979.
[6]吴三多,赖登燡,温宽和,等.北方地区窖泥退化原因及管理养护的研究[J].酿酒科技,2014(9):71-74.
[7]王明跃,张文学,王海英,等.不同窖龄窖泥细菌的系统发育多样性分析[J].食品科学,2013(11):177-181.
[8]王涛,田时平,赵东,等.宜宾浓香型白酒窖泥中细菌的系统发育多样性[J].食品与发酵工业,2011,37(10):11-18.
[9]刘念,刘绪,张磊,等.浓香型白酒糟醅中真菌群落的研究[J].食品与发酵工业,2011,47(2):28-31.
[10]罗惠波,甄攀,黄治国,浓香型白酒窖池细菌群落[J].微生物学通报,2010,37(11):1621-1627.
[11]黄治国,甄攀,罗惠波,浓香型白酒窖池细菌群落[J].西南大学学报:自然科学版,2010,32(12):91-96.
[12]邓依,唐云容,张文学.16S-23S rRNAITS-AFLP指纹图谱分析在白酒窖泥原核微生物多样性分析中的应用[J].酿酒科技,2010(3):46-48.
[13]Lauber C L,Hamady M,Knight R,et al.Pyrosequencingbased assessment of soil pH as a predictor of soil bacterial community structure at the continental scale[J].Appl Environ Microbiol,2009,75(15):5111-5120.
[14]曾祥勇,董雅舒,胡贝,等.不同年份窖泥细菌16S rDNA系统发育分析[J].四川大学学报,2014(9):71-74.
[15]黄永光,黄平,涂华彬.窖泥微生物总DNA的提取纯化研究[J].酿酒科技,2004(3):41-42.
[16]Westerholm M,Roos S,SchnurerA.Syntrophaceticus schinkii gen.nov.,sp.nov.,an anaerobic,syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter[J].FEMS Microbiol Letters,2010,309(1):100-104.
[17]Kobayashi T,Yan F,Takahashi S,et al.Effect of starch addition on the biological conversion and microbial community in a methanol-fed UASB reactor during long-term continuous operation[J].Bioresource Technol,2011,102:7713-7719.
[18]Good I J.The population frequencies of species and the estimation of population parameters[J].Biometrika,1953,40:237-264.
[19]陕小虎,敖宗华,周健,等.浓香型白酒窖泥原核微生物DGGE电泳条件的优化[J].酿酒科技,2011(1):37-40.
[20]Lee S H,Kang H J,Lee Y H,et al.Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters[J].J Environ Monit,2012,14(7):1893-1905.
[21]Deng B,Shen C H,Shan X H,et al.PCR-DGGE analysis on microbial communities in pit mud of cellars used for different periods of time[J].Journal of the Institute of Brewing,2012,118(1):120-126.
[22]Thomton R H.Fungi occurring in mixed oakwood and heath soil profiles[J].Trans Brit Mycol Soc,1956,39:484-494.
[23]Widden P.Fungal communities in soils along an elevation gradient in northern England[J].Mycologia,1987,79:298-309.
[24]Gochenaur S E.Fungi of a Long Island oak-birch forest II. Population dynamics and hydrolase patterns for the soil penicillia[J].Mycologia,1984,76:218-231.
[25]Dobranic J K,Zak J C.Amicrotiter plate procedure for evaluating fungal functional diversity[J].Mycologia,1999,91:756-765.
[26]Mallik MAB,Elroy L.Relation between soil fungi and seed plant in three successional forest communities in Oklahoma[J]. Bot Gaz,1966,127:3120-3127.
[27]Hawksworth D L.The fungal dimension of biodiversity: magnitude,significance,and conservation[J].Mycol Res,1991,95:641-655.
[28]邹莉,唐庆明,王轶.落叶松,樟子松纯林及混交林土壤微生物的群落分布特征[J].东北林雪大学学报,2010,38(11):63-79.
[29]巨天珍,陈源,常成虎,等.天水小陇山红豆杉林土壤真菌多样性及其与生态因子的相关性[J].环境科学研究,2008,21(1):129-131.
[30]Dennis P G,Rushton S P,Newsham K K,et al.Soil fungal community composition does not alter along a latitudinal gradient through the maritime and sub-Antarctic[J]. Ungalecology,2012,5:403-408.
[31]Kasel S,Bennett LT,Tibbits J.Land use influences soil fungal community composition across central Victoria[J].Soil Biology&Biochemistry,2008,40:1724-1732.
[32]吴敏娜,张惠文,李新宇,等.土壤抑真菌作用与细菌群落结构的关系[J].应用生态学报,2008,19(7):1574-1578.
[33]刘绪,张磊,王超凯,等.窖泥中优势丝状真菌的分布规律及分类鉴定[J].食品与发酵工业,2012,48(1):16-19.
Differences of Fungal Communities in Nongxiang Baijiu Pit Mud before and after the Deterioration
YU Chuntao1,LIU Chao1,CHEN Ruiling1,ZHU Fenglin1and LIU Zhenjiang2
(1.Cangzhou Medical College,Cangzhou,Hebei 061001;2.Nikeng Distillery Co.Ltd.,Shijiazhuang,Hebei 054000,China)
Pit mud deterioration is a major problem for distilleries in North China.In this study,total DNA of fungi in pit mud was extracted directly and high throughput sequencing method was used to amplify 18S rDNA and to construct gene library to analyze the composition of fungal communities in pit mud before and after the deterioration.The results suggested that,there were 9737 sequences belonging to 2 phylums before the deterioration,and the dominant fungi included Microascus(74.21%),Penicillium(10.03%),Pseudogymnoascus(9.99%),etc.There were 78322 sequences belonging to 19 phylums after the deterioration,and the dominant fungi included Penicillium(76.56%),Pseudogymnoascus(6.73%),Inonotus(5.96%),Inocybe(2.93%),etc.There were significant differences in fungi varieties and abundance before and after the deterioration.This study provided a theoretical base for the comprehensive understanding of pit mud deterioration.
pit mud;deterioration;fungal community;analysis of the difference;high throughput sequencing
TS262.3;TS261.4;TS261.1
A
1001-9286(2016)10-0040-05
10.13746/j.njkj.2016159
2016-05-09;
2016-06-16
于春涛(1979-),男,河北省沧州市人,硕士研究生,主要从事微生物发酵研究。
优先数字出版时间:2016-07-11;地址:http://www.cnki.net/kcms/detail/52.1051.TS.20160711.1103.005.html。
