丙烷CVI工艺热解炭沉积非均相反应动力学模拟
2016-10-31汤哲鹏李爱军张中伟白瑞成王俊山任慕苏
汤哲鹏, 徐 伟, 李爱军, 张中伟, 白瑞成, 王俊山, 任慕苏
(1.上海大学 材料复合及先进分散技术教育部工程中心,上海200072;2.航天材料及工艺研究所 先进功能复合材料技术国防科技重点实验室,北京100076)
丙烷CVI工艺热解炭沉积非均相反应动力学模拟
汤哲鹏1,徐伟1,李爱军1,张中伟2,白瑞成1,王俊山2,任慕苏1
(1.上海大学 材料复合及先进分散技术教育部工程中心,上海200072;2.航天材料及工艺研究所 先进功能复合材料技术国防科技重点实验室,北京100076)
分别运用总括非均相反应机理和详细非均相反应机理,结合均相反应机理(包括285种气相组分,1 074个气相可逆基元反应)来模拟C3H8在CVI工艺条件下炭纤维表面热解炭的沉积过程,进而对实验中的气相组分和热解炭的形成过程进行预测。总括非均相反应机理对炭沉积反应进行了简化处理,气相中的烃组分直接在表面脱氢沉积为热解炭;而详细非均相反应机理则利用表面基元反应来描述热解炭沉积过程,包括66种表面组分和250个表面基元反应。本文以C3H8为炭源,N2为稀释气体,温度1 173~1 323 K、低压(2.6 kPa)和滞留时间为0.5~4 s条件下的连续搅拌釜反应器为模型进行模拟,气相组成和沉积动力学两方面的预测与实验结果都较好吻合。计算表明在该设定条件下热解炭的前驱体主要为不饱和小分子(C2H2和C2H4)和甲基,进而利用这些组分定量解释热解炭的沉积动力学。
模拟; 热解炭; 表面动力学; 丙烷; 化学气相渗透
1 前言
20世纪60年代人类制备出C/C复合材料,其中由烃类到C/C复合材料的CVI工艺引起人们极大的兴趣[1]。烃类如丙烷在高温900~1 500 K、低压1~50 kPa条件下热解,同时在多孔预制体的炭纤维表面沉积热解炭成为基体。除热解炭外,热解还产生大量的气相组分如H2、各种轻质烃、芳香烃和稠环芳香烃[2-5]。
用气态烃制备热解炭时,由于热解炭前驱体在预制体孔隙中的传质受阻导致了较长的致密化周期和较低的沉积速率,使得生产成本颇高。深入研究前驱体热解过程中涉及的反应和热解炭沉积机理有利于帮助我们优化C/C复合材料生产工艺和降低生产成本[6,7]。
要弄清热解炭的沉积机理,首先需要详细地描述丙烷的气相热解反应。为此,笔者已提出了一个包括285种气相组分(分子或自由基)和1 074个基元反应详细的气相反应机理[8]。该机理能预测气相中从CH4到芘等主要产物的形成。其次,需要详细地描述热解炭的沉积反应。关于热解炭沉积的描述,Becker和Hüttinger使用“表面活性位”上的反应来描述[9-11],热解炭的沉积反应被认为是非均相反应,经吸附、表面反应和解吸附三个过程。他们提出了一个可以描述C2类烃的炭沉积动力学的简化模型,使用集总方法将大量的气相组分集成处理成三类,如C2、C4和C6。涉及的表面反应速率常数基于Langmuir-Hinshelwood动力学原理,通过拟合实验结果来估计。此方法在考虑复杂气相化学反应的基础上,首次成功地使用L-H动力学描述了热解炭的沉积。
Ziegler等利用总括反应描述热解炭的沉积反应[12],避免了使用表面组分来描述热解炭表面。总括沉积反应机理将热解炭的沉积反应当成是气相组分直接沉积生成热解炭的不可逆气相反应。例如从气相组分CnHm到热解炭的沉积写成以下经验形式:
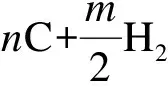
(1)
式中,“C”是组成热解炭层的碳原子,总括沉积反应可以写入气相反应机理中,总括反应的速率常数则通过拟合模拟结果与CVD实验结果得到,使用热解炭的虚拟浓度来表征沉积速率。当考虑传质现象的影响时,一般也是使用总括沉积反应机理来描述热解炭沉积的表面反应。Vignoles等[13-15]为了确定炭沉积期间气相扩散和沉积动力学的重要性,提出了一种模拟方法,该模型中考虑了C2H2、苯和萘的沉积。李爱军等[16]探索从甲烷到热解炭的CVI过程时采用了类似方法,提出并成功证实热解炭主要由乙烯、乙炔和苯沉积构成的动力学模型,其原始的表面反应动力学数据是通过拟合模拟结果与CVD的实验结果得到。Lacroix等[17]首次提出了基于基元反应的详细表面动力学机理来描述热解炭的沉积反应。对于热解炭沉积涉及到的吸附、表面反应和解吸附等全部使用基元反应步骤来表述。
本文分别利用总括反应和详细的基元反应来描述热解炭沉积时的非均相反应,从而对丙烷CVI的表面反应进行动力学模拟。在所选条件下,总括反应机理侧重考虑C2H4、C2H2、苯、菲和蒽5种潜在热解炭前驱体的沉积。详细非均相反应机理则考虑气相中主要的自由基和分子在热解炭边缘的“Zig-zag”和“Armchair”活性位上发生吸附、表面反应和解吸附等基元步骤。最后,通过热解炭沉积反应机理与气相热解反应机理间的耦合,对丙烷CVI工艺的热解炭沉积过程进行了数值模拟。
2 模拟方法
2.1机理
热解炭的沉积机理有生长机理和形核机理两种。在CVI条件下,复杂的均相热解反应和非均相沉积反应相互竞争或相互作用,因多孔预制体极高的比表面,使得气相大组分生成反应受到明显抑制,故热解炭的沉积以小分子的生长机理为主。本文重点研究沉积初期具备较大孔隙时,忽略传质限制的情况。
本文使用的气相热解反应机理源自编辑文献中发表的基元反应。此反应机理由285种组分1 074个基元反应,其中1 000个反应为可逆反应。该反应机理的参考文献、所有组分的热力学数据、组分名称简写和相应化学式等详细信息见文献[8]。
动力学数据处理时,该气相反应机理仅给出基元反应动力学参数中的正向反应速率常数,其中绝大部分反应速率常数服从Arrhenius定律,而任一基元反应的逆向反应速率常数则基于相关组分的热力学数据,通过以下公式来计算:
(2)
(3)
(4)
式中,kr和kf分别为正逆反应速率常数,KC为反应的浓度平衡常数,c0标准浓度,ΔRG0标准反应吉布斯自由能,ΔRv为反应的化学计量之和,P0标准压力,R理想气体常数,T开氏温度。对那些压力敏感的基元反应,其反应动力学参数需要用Troe参数来表示。还有些反应有第三体参加,因CVD/CVI工艺过程中轻质烃一般在低压条件下热解,所以在研究轻质气态烃分压对主要组分的影响时,使用第三体和Troe参数描述反应速率常数显得十分重要。
目前,由于尚不清楚在热解炭沉积表面上发生了什么反应,所以对于何种组分沉积成热解炭这个问题,我们需要在两种假定中作出选择。一种假定认为参与沉积的气相组分会消失或数量很少,没有参与沉积的气相组分数量多;另一种假设则认为数量多的气相组分参与热解炭沉积[18]。本文选择后一种假定,在修正Ziegler等[12]和Lacroix等[17]提出的反应机理基础下,分别使用了总括非均相沉积反应机理和详细的基元反应机理。
2.1.1. 总括表面反应机理
总括反应机理着重考虑两种类型的总括沉积反应:一类是包含C2H2这样的不饱和小分子气相组分的反应;另一类是包含大分子(≥C6)的反应。事实上,丙烷热解产物中不饱和的小分子组分是主要的产物,它们的摩尔数和热解炭相近。并且这些不饱和小分子多数含有双键或三键,它们会参与到热解炭的生长反应中。特别是乙炔,它的加成作用非常重要。气相中大组分同小组分相比数量较低,但它们也很有可能参与热解炭的沉积,因为它们的结构和热解炭非常相近,都是碳原子结合形成的六元环碳平面。气态烃“CnHm”的总括沉积反应见公式(1)。假定热解炭沉积速率相对气态烃“CnHm”为一级反应,则沉积速率表示为:
(5)
式中,k为速率常数(s-1),[CnHm]是热解炭前驱体组分的实验浓度(mol·cm-3),n是气态烃“CnHm”中碳原子数,r是体积沉积速率(mol·cm-3·s-1)。实验确定热解炭的质量沉积速率rmass(g·s-1),则根据质量沉积速率可得体积沉积速率r和速率常数k:
(6)
(7)
式中,MC为碳原子的摩尔质量,V为反应器体积。假定速率常数在所研究的温度范围内符合Arrhenius定律,则可用直线拟合实验测定速率常数的自然对数值与温度倒数的关系从而得出总括反应速率常数的Arrhenius参数。由于不是基元反应,因此总括反应的动力学参数只适用于所给实验条件。将带有Arrhenius参数的总括反应写入丙烷均相热解的机理中,就可以像模拟均气相反应那样进行热解炭沉积的模拟,其中热解炭被假定为虚拟气相组分。根据输入的参数可以计算出包括热解炭在内所有组分的摩尔分数。假定热解炭在反应器中积累,则模拟的热解炭质量可以计算:
(8)
式中,m为热解炭质量(g),XC为热解炭摩尔分数,V反应器体积(m3),τ气体滞留时间(s),t实验沉积时间(s)。
特别需要注意只有热解炭的虚拟分压比总压低,这种总括方法才是有效的。事实上,具有双键或三键的脂肪族烃数量较大,它们也可能反应导致热解炭形成。模拟热解炭沉积速率时还需要考虑从C2H2到C4H2等不饱和组分共沉积效应,则体积沉积速率r表示为:
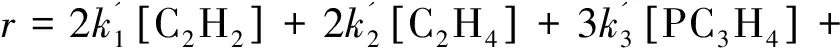
(9)
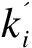
2∑C2摩尔分数≫3∑C3摩尔分数+4∑C4摩尔分数
(10)
因此,热解炭体积沉积速率可以简化成:
(11)
(12)

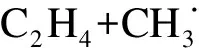
E=32.2 kJ/mol
(13)
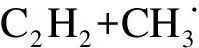
E=32.2 kJ/mol
(14)
(15)
通过实验确定体积沉积速率“r”、在不同温度下乙炔和乙烯的浓度,就可以算出相应温度下的总括反应速率常数k。基于Arrhenius公式拟合k与温度的关系求出乙炔和乙烯共沉积时的总括反应动力学参数如下所示:
C2H2⟹2C+H2,A=7.7×1010s-1,
E=281.0 kJ/mol
(16)
C2H4⟹2C+H2,A=4.1×1010s-1,
E=281.0 kJ/mol
(17)
2.1.2详细表面反应机理
基于基元反应步骤的详细表面反应机理的建立,首先需要对热解炭表面进行描述。忽略预制体内纤维之间的接触面积,将预制体假想成一根单独圆柱形纤维,则根据纤维的直径d、密度ρ、质量m可得出纤维假想长度L:
(18)
因此气体与炭纤维表面的接触面积可以估计为S=πdL,初始孔隙率可以用以下公式 计算:
(19)
对表面反应进行计算时需要知道表面活性位浓度[19],化学吸附就发生在表面活性位上。目前,这个参数尚未通过实验进行确定,并且也无法通过考虑石墨的实际密度来获得,因为只有位于基本结构单元边缘位置的碳原子才代表活性位。本文基于Lacroix等[17]提出的估计方法,使用含有225个芳香环的正方形稠环芳香烃来代表热解炭上层的一部分(图1)。

图 1 表面活性位浓度估计示意图
在这部分平面上同时发生着几个基本结构单元的侧向生长。表面活性位的数量是位于基本结构单元边缘上的碳原子,本文表面活性位浓度为8.7×1014个cm-2。
由于基本结构单元的侧向生长在化学上类似于气相芳香烃的生长,因此认为表面基元反应与气相热解中的脱氢、不饱和组分加成、β剪切、ipso加成、起始和终止等反应中涉及到的基元反应类似。非均相反应被认为是发生在基本结构单元边缘上的“Zig-zag”和“Armchair”活性位上见图2。
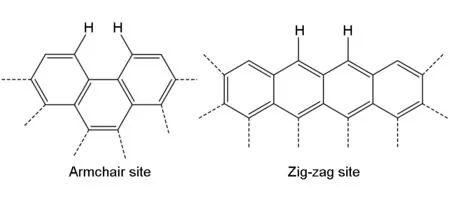
图 2 “Armchair”和“ig-zag”表面位点
要将“Armchair”活性位转化成6个碳的芳香环至少需要2个碳原子,而“Zig-zag”则至少需要3个碳原子。Lacroix等人提出的机理中考虑了“Zig-zag”和“Armchair”活性位上的热解炭沉积,并假定两种活性位各占一半。不考虑热解炭在缺陷和杂质上的沉积,也不考虑五元环和七元环的形成,因为它们只对热解炭的织构有较大影响,热解炭的微观结构不在讨论范畴之内。
详细的非均相反应机理主要包括3种类型的反应,双分子气相/表面反应、单分子表面反应和双分子表面反应。双分子气相/表面反应的形式写为:A(S) + B(G) →产物,相应的反应速率表达式为:
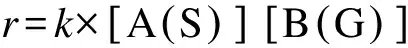
(20)
式中,r为反应流速率(mol cm-2s-1),k为双分子反应速率常数(cm3mol-1s-1),[A(S)]为A(S)表面组分浓度(mol cm-2),[B(G)]为气相组分B(G)的浓度(mol cm-3)。单分子表面反应的形式为:A(S) →产物,相应的反应速率表达式为:
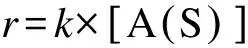
(21)
式中,k的单位为s-1。双分子表面组分反应的形式写为:A(S) + B(S) → 产物,相应的反应速率表达式为:

(22)
动力学速率常数单位为cm2mol-1s-1,并且反应流速率仍以(mol cm-2s-1)为单位,而这实际上与使用气相原型反应来估计表面动力学参数相矛盾。因此借鉴Lacroix等[17]的方法确定温度因子和活化能,再通过拟合实验结果修改指前因子。
关于详细表面基元反应机理,目前主要借鉴已有的成焦机理、炭黑和金刚石薄膜的生成机理[9,20-23]。在这些工作中表面基元反应动力学数据以选定的“原型气相反应”为依据,依照量子化学的碰撞理论加以修正。本文借鉴了金刚石薄膜制备工艺表面基元反应机理开发中使用的“原型气相反应”方法。其中,Lacroix等[17]采用原型气相基元反应的方法来估计表面基元反应动力学参数。该方法将与表面基元反应在化学上相似的气相基元反应作为“原型反应”,且假定它们的动力学参数都服从Arrhenius定律。表面反应的指前因子和活化能与气相原型反应是相同的,对于有一些表面基元反应,则考虑用气相原型和表面基元反应中的反应路径退化因子来修正指前因子。由于基本结构单元包含相当高数量的炭环,所以理想的气相原型反应中应该包含炭环数量相对较高的烃。但大量出版文献中的详细气相反应机理中,都认为涉及稠环芳香烃的化学反应的动力学参数和基于苯和甲苯等单环芳香烃的类似反应的动力学参数相同[24]。同时,一些学者使用ab initio方法研究了芳香烃的大小对一些反应的动力学参数的影响,例如在1 273 K,H原子夺去稠环芳香烃(PAH)边缘位置上H的反应的速率参数对于直至7个芳香环的PAH几乎相同[25]。相似地基于ab initio计算表明,对于苯、萘、蒽、并四苯和并五苯通过甲基的脱氢反应,它们的速率常数是相同的[26]。乙炔在甲基上的加成也得到相似的结果[27]。因此,可以将包含苯及其衍生物的气相反应作为原型反应用来估计表面反应的动力学参数。
Lacroix等认为六个碳原子芳香环的形成从氢化的折椅位点和之字位点开始。首先是氢化的折椅位点和之字位点脱氢生成活性位;其次是活性位吸附或解吸附气相中的自由基和分子;最后是发生沉积反应形成固体炭。基于实验结果,本文只考虑主要的气相分子烃如乙炔、乙烯、丙炔、丙二烯、苯、萘、苯乙炔和苯乙烯,自由基如H、甲基、乙烯基,乙基、乙炔基、乙烯基乙炔基、苯亚甲基和茚基等物质参与的表面非均相反应。
2.2CVI反应器建模
采用Ziegler等[28]在丙烷热解实验中所使用的喷气式搅拌CVD反应器。该反应器由四个喷嘴同时沿4个方向喷出的气体来搅拌混合,从而保证气体在整个反应器内完全均匀混合,这样在整个反应器内气体组成相同,故可以精确地知道在热解炭沉积条件下某时刻的气相组成。因此,可直接理想化成具有与实际反应器相同体积的零维球形连续搅拌釜反应器(CSTR)模型,并结合丙烷裂解反应机理,建立起丙烷CVI工艺热解炭非均相沉积的0D反应动力学模型,其控制方程包括:
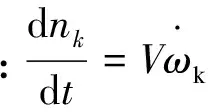
(23)
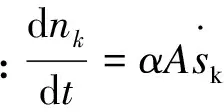
(24)
理想气体状态方程: pV=nRT
(25)
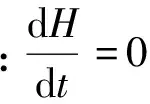
(26)
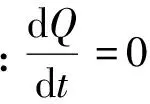
(27)
2.3实验数据
验证本文零维CVI反应流动力学模型的实验数据来自Lacroix等[17]和Ziegler等[12]自行设计的气体喷嘴搅拌连续搅拌釜反应器实验,热解炭沉积的工艺条件为:总压2.7 kPa,丙烷分压0.35 kPa,N2为稀释气体,气体滞留时间为1 s,沉积温度为1 173~1 298 K;沉积时间为90 min,沉积基体为环形的炭纤维预制体,其直径为7.5 μm,质量约为1.3 g。选择较短沉积时间可避免前驱体在预制体中沉积动力学受传质的限制。沉积期间产生的气相组分使用气相色谱仪来分析,热解炭的质量则用微重仪来测量。
Lacroix等[17]用相同的反应器在总压2.6 kPa,丙烷摩尔分数1/9,N2为稀释气体,沉积温度为1 173~1 323 K,滞留时间为0.5~4 s,沉积时间为90 min的条件下进行了沉积实验。同时,为研究预制体对气相组成的影响,分别在有炭纤维预制体和无炭纤维预制体的反应器中进行了沉积实验。
3 结果与讨论
3.1气相组成
在总括反应机理中,本文只考虑小分子乙炔和乙烯的共沉积,其中乙烯和乙炔共沉积的动力学参数通过实验拟合得到,本文采用Ziegler等[12]发表的实验数据。结合均相反应机理和乙烯与乙炔的共沉积反应,计算丙烷均相热解时气相产物浓度随温度的分布函数图,并将全混反应器的模拟计算结果同实验结果进行比较,见图3。
总体上,计算结果与实验结果吻合。事实上,热解炭的生成会导致乙炔和乙烯有较大的消耗,故乙烯和乙炔的预测值要比丙烷纯均相热解时的预测值低,但乙烯和乙炔的浓度并没有因共沉积沉炭致其预测值过低。相反,在考虑表面反应后,乙烯和乙炔浓度的计算值更接近实验值,说明低温低压条件下,丙烷热解过程中炭的沉积反应主要是乙烯和乙炔的共沉积。由于乙炔和乙烯的共沉积,可以预想有炭纤维时H2浓度的预测值会上升。当引入预制体时C3浓度下降,而图3未能预测出这种变化,丙炔和丙二烯的浓度计算值在低温时比丙烷纯均相热解时的预测值低,而在高温时则相反。考虑乙烯和乙炔共沉积后,苯浓度的计算值与纯气相热解情况几乎相同,这主要是因为苯存在多条生成路径,同时又缺少苯的消耗反应,使得浓度偏高,异于实验结果。
在低压条件下,考虑表面反应(乙烯和乙炔共沉积)后,气相产物浓度的预测值更接近实验值。但实验和计算表明,与丙烷纯均相热解相比,丙烷非均相热解的气相产物浓度变化不大。所以,可使用丙烷均相热解时的气相产物组成来近似预测丙烷在CVD或CVI条件下发生热解炭沉积时的气相产物组成。丙烷在低压CVI条件下热解时,预制体的孔隙(比表面积)对气相产物组成的影响可以忽略,但孔隙(孔隙当量半径)对气相产物的传质有重要的影响,这将在后续工作中重点研究。
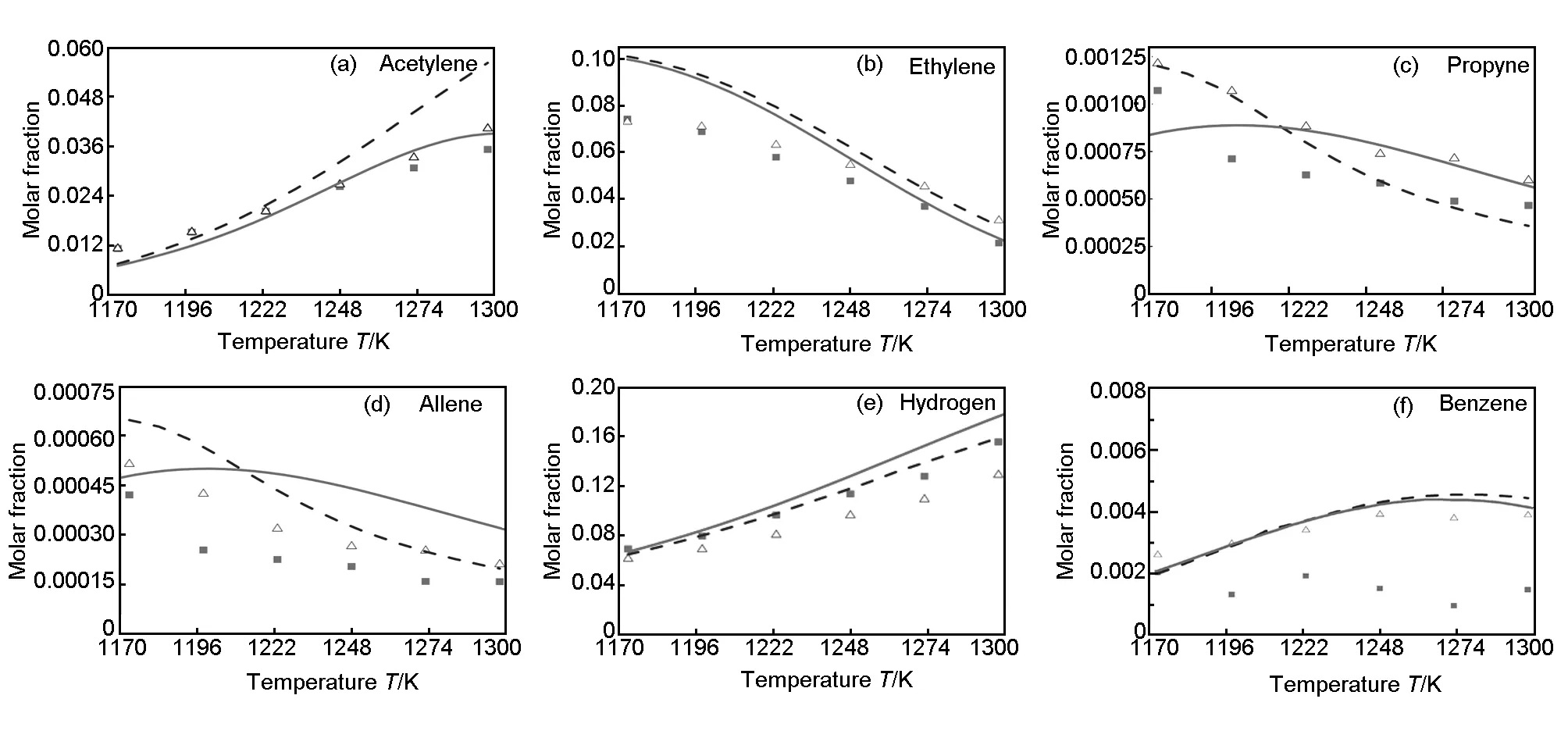
图 3 乙烯和乙炔共沉积时气相组分随温度分布的计算结果(曲线)与实验结果(符号)相比较.(△ , 无表面反应的实验结果; ■, 有表面反应的实验结果; ┄, 无表面反应的计算结果; —, 有表面反应的计算结果)
在详细表面基元反应机理中,本文考虑主要的烃分子和自由基在“Zig-zag”和“Armchair”活性位的吸附和热解炭沉积,并结合均相反应机理,以全混反应器模型来计算丙烷CVI热解初期主要气相组分随温度和滞留时间的变化,并将计算结果同实验结果进行比较,实验数据来自文献[17](图4)。
总体上,计算结果与实验结果吻合。其中,对C2H2的浓度随温度的分布预测较为准确,H2的预测值稍高,而对苯和萘的浓度随温度变化的预测值与实验值偏差较大。但计算结果仍能预测浓度随温度的变化趋势。与丙烷的纯均相热解相比,引入预制体后H2的浓度升高,这是由除氢和脱氢反应引起的;而对其他烃类产物来说,引入预制体后其浓度降低,其原因主要有两方面:一是由沉积反应引起烃类大量消耗;二是由前驱体消耗所引起的较小生成速率。这些现象皆通过该详细反应动力学模型得到验证。
由图4可知,小分子组分的摩尔分数能够准确地通过该详细反应动力学模型来预测,实际上模拟曲线和实验曲线十分接近。相反,芳香烃的摩尔分数预测值都偏高。其原因有两方面:首先是由于在反应机理中缺少对所考虑组分的消耗反应;其次是由反应机理中涉及的许多大分子基元反应的动力学参数不准确性。
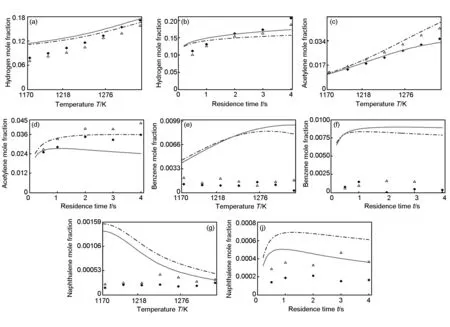
图 4 主要产物的摩尔分数随温度和滞留时间的分布函数图(△: 无预制体的实验结果, ◆: 有预制体的实验结果, —: 有预制体的计算结果, ┄: 无预制体的计算结果)
3.2热解炭沉积
本文侧重研究低温低压条件下,丙烷化学气相渗透初期的非均相反应动力学,故不考虑传质受限问题。基于丙烷均相反应的总括表面沉积反应机理,只考虑乙烯和乙炔的共沉积热解炭的沉积动力学,将热解炭的沉积看作乙烯和乙炔两种物质不可逆地沉积成假想的“气态热解炭”。
结合均相反应机理和乙烯与乙炔的共沉积反应,基于全混反应器模型来计算丙烷均相热解时“气态热解炭”随温度的分布函数,运用公式(7)计算出热解炭的质量随温度的变化函数。将计算的热解炭质量同Ziegler[12]等所报导的实验结果相比较见图5。本文研究条件下,丙烷CVI初期热解炭的沉积量随温度升高呈指数增加。由此说明,在低温低压条件下,丙烷CVI热解炭的沉积源自乙炔和乙烯等小分子。

图 5 乙烯和乙炔共沉积时热解炭的质量随温度变化的计算结果(曲线)与实验结果(符号)的比较
结合详细表面基元反应机理和均相热解反应机理,计算丙烷CVI工艺初期热解炭沉积速率随温度和滞留时间的分布函数,计算结果较之Lacroix[17]的实验结果见图6。热解炭的速率表示为化学反应流体速率,当温度升高150 K时,相应热解炭速率变为原来6倍。该详细化学反应动力学模型能够很好地预测温度和滞留时间对丙烷CVI初期热解炭沉积速率的影响。热解炭沉积速率随温度呈指数增长,随滞留时间呈指数下降。温度不仅影响热解炭沉积反应速率常数,而且改变热解炭沉积前驱体组成。滞留时间延长有利于生成大分子组分,虽然单位表面积大分子的成炭速率比小分子高,但大分子扩散系数小传质较困难,故延长滞留时间会降低热解炭沉积速率。
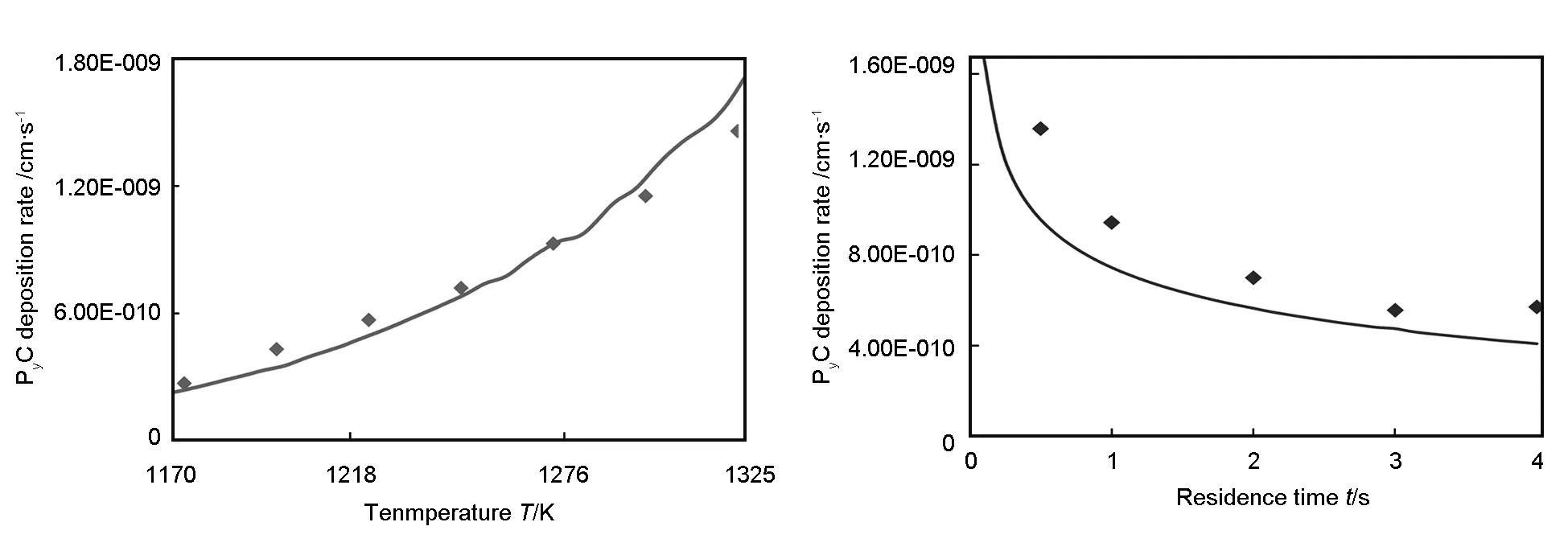
图 6 热解炭沉积速率随温度和滞留时间分布的计算结果(曲线)与实验结果(符号)的比较.
4 反应流速率分析
在温度1 273 K,滞留时间1 s,总压为2.6 kPa,炭纤维与气体的接触面积为3 960 cm2,入口丙烷摩尔比率为1/9,N2为稀释气体等条件下,基于全混反应器模型,对热解炭的沉积进行反应流速率分析见图7。
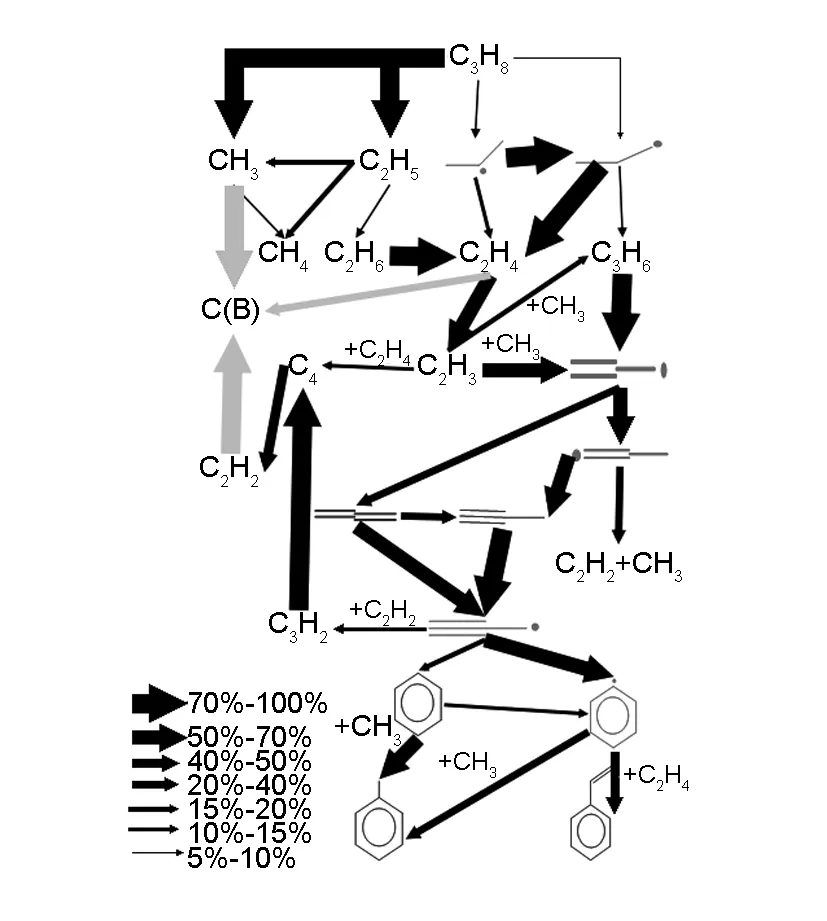
图 7 丙烷热解反应流速率分析(T=1273 K,滞留时间=1 s).
由图7可知,存在表面反应促使丙烷通过单分子反应裂解成甲基和乙基,因为甲基在表面反应中非常活跃,消耗的速度较快,从而有利于丙烷单分子裂解,它通过夺氢反应使表面解吸附H而生成活性位。在计算结果中,发现甲基的浓度非常低。另外表面反应只对乙烯、乙炔、甲烷和甲基等小分子组分的浓度有较大影响。这说明在此条件下,主要是由小分子吸附反应沉积成炭。由此可知,在本文研究条件下,表面反应使丙烷的热解反应路径变得更为简单,因为表面反应在不停地消耗热解中间产物,抑制了气相反应。“Armchair”活性位主要通过吸附CH4和H2上的H而被消耗掉,而其主要通过气相中的H、CH3等活性自由基的除氢反应生成;“Zig-zag”活性位主要通过吸附CH3和H2与CH4上的H被消耗掉,而其也是主要通过气相中的H、CH3等活性自由基的除氢反应生成。
在致密化初期,炭纤维表面存在活性位,在此条件下块体炭主要通过以下两条途径累计:一是“Zig-zag”活性位吸附CH3和CH4与H2上的H累计,二是“Zig-zag”活性位吸附物之间发生表面反应。26%的热解炭是由丙炔吸附在“Zig-zag”活性位上沉积而成的,CH3和C2H4或C2H2依次吸附在“Zig-zag”活性位发生沉积反应,贡献约22%的热解炭,其余可来自3个CH3依次吸附在“Zig-zag”活性位上沉积。由此可见热解炭主要由小的不饱和分子(乙烯、乙炔和丙炔)在“Zig-zag”位点上沉积而成。在相同条件下,Lacroix在文献中指出在“Armchair”位点上,热解炭主要为乙炔和乙烯等不饱和小分子和甲基的沉积。他提到33%的热解炭沉积速率得益于甲基在“Armchair”位点上的沉积,而乙烯和乙炔的沉积分别贡献12%和20%。本文热解炭沉积主要为乙烯、乙炔、丙炔和甲基在“Zig-zag”位点上沉积,而在“Armchair”的沉积却非常小,考虑到“Armchair”位点存在较大的空间位阻,并且表面基元反应的动力学参数的不准确给计算结果带来较大的不可预测性,这些都可能导致出现这种结果。
5 结论
分别使用两种表面反应机理描述丙烷在CVI条件下热解炭沉积时发生的非均相反应。基于已经建立的丙烷均相热解反应机理,首先考虑热解炭从乙炔和乙烯共沉积的总括表面反应机理,结合全混反应器模型,较为准确地预测了丙烷在温度1 173-1 298 K,分压0.35 kPa,总压2.7 kPa,N2为稀释气体,滞留时间1s等条件下热解炭的沉积动力学;其次,使用基元反应步骤的非均相表面反应机理并结合全混反应器模型也较为准确地预测丙烷在温度1 173~1 323 K,分压0.26 kPa,总压2.6 kPa,N2为稀释气体,滞留时间1 s等条件下热解炭的沉积动力学。
计算和实验都表明,在本文研究的条件下,丙烷在有非均沉积情况下的热解成分和仅存在均相热解时主要气相产物的变化不大。因此,丙烷用作CVD或CVI时,对于小型的多孔预制体可使用丙烷均相热解时的气相产物组成近似表达热解炭沉积时的气相产物组成。但另一方面,表面反应促使丙烷裂解起始反应几乎全部转向单分子分解反应。在本文研究条件下,热解炭主要由乙烯和乙炔等不饱和小分子和甲基的沉积而来。
未来工作在优化反应机理的同时,需要模拟在CVI条件下的真实传质和传热过程。在CVI过程中存在明显扩散限制问题,因此需要模拟预制体随致密化进行孔隙率和孔隙形貌的演变过程,另外还要估计气相组分和表面组分的扩散动力学参数。除此之外,需要正确模拟实际反应器的温度场和流场,使之更接近真实的反应条件。
[1]Savage G. Carbon-Carbon Composites[M]. London: Chapman & Hall, 1993.
[2]Norinaga K, Deutschmann O. Detailed kinetic modeling of gas phase reactions in the chemical vapor deposition of carbon from light hydrocarbons[J]. Ind Eng Chem Res, 2007, 46(11): 3547-3557.
[3]Norinaga K, Janardhanan V M, Deutschmann O. Detailed chemical kinetic modeling of pyrolysis of ethylene, acetylene, and propylene at 1073-1373 K with a plug-flow reactor model[J]. International Journal of Chemical Kinetics, 2008, 40(4): 199-208.
[4]Norinaga K, Deutschmann O, Hüttinger K J. Analysis of gas phase compounds in chemical vapor deposition of carbon from light hydrocarbons[J]. Carbon, 2006, 44(9): 1790-1800.
[5]Norinaga K, Deutschmann O, Saegusa N. Analysis of pyrolysis products from light hydrocarbons and kinetic modeling for growth of polycyclic aromatic hydrocarbons with detailed chemistry[J]. J Anal Appl Pyrolysis, 2009, 86(1): 148-160.
[6]Ziegler I, Fournet R, Marquaire P M. Pyrolysis of propane for CVI of pyrocarbon Part I. Experimental and modeling study of the formation of toluene and aliphatic species[J]. J Anal Appl Pyrolysis, 2005, 73(2): 212-230.
[7]Ziegler I, Fournet R. Pyrolysis of propane for CVI of pyrocarbon Part II:Experimental and modeling study of ployaromatic species[J]. J Anal Appl Pyrolysis, 2005, 73(2): 231-247.
[8]徐伟, 张中伟, 白瑞成, 等.丙烷化学气相沉积均相热解反应动力学模拟[J]. 新型炭材料, 2014, 29(01): 67-77.
(XU Wei, ZHANG Zhong-wei, BAI Rui-cheng, et al. Kinetic modeling of gas-phase reactions for CVD from propane[J]. New Carbon Materials, 2014, 29(01), 67-77.)
[9]Becker A, Hüttinger K J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon-II pyrocarbon deposition from ethylene, acetylene and 1,3-butadiene in the low temperature regime[J]. Carbon, 1998, 36(3): 177-199.
[10]Becker A, Hüttinger K J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon-III pyrocarbon deposition from propylene and benzene in the low temperature regime[J]. Carbon, 1998, 36(3): 201-211.
[11]Becker A, Hüttinger K J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon-IV pyrocarbon deposition from methane in the low temperature regime[J]. Carbon, 1998, 36(3): 213-224.
[12]Ziegler I, Fournet R, Marquaire P M. Pyrolysis of propane for CVI of pyrocarbon: Part III: Experimental and modeling study of the formation of pyrocarbon[J]. Journal of Analytical and Applied Pyrolysis, 2007, 79(1-2): 268-277.
[13]Descamps C, Vignoles G L, Féron O. Correlation between homogeneous propane pyrolysis and pyrocarbon deposition[J]. J Electrochem Soc, 2001, 148(10): 695-708.
[14]Descamps C, Vignoles G L, Féron O, et al. Kinetic modeling of gas-phase decomposition of propane: Correlation with pyrocarbon deposition[J]. Journal de Physique IV, 2001, 11(PR3): 101-108.
[15]Vignoles G L, Gaborieau C, Delettrez S. Reinforced carbon foams prepared by chemical vapor infiltration: A process modeling approach[J]. Surface and Coatings Technology, 2008, 203(5-7): 510-515.
[16]Li A, Norinaga K, Zhang W. Modeling and simulation of materials synthesis: Chemical vapor deposition and infiltration of pyrolytic carbon[J]. Composites Science and Technology, 2008, 68(5): 1097-1104.
[17]Lacroix R, Fournet R, Ziegler I, et al. Kinetic modeling of surface reactions involved in CVI of pyrocarbon obtained by propane pyrolysis[J]. Carbon, 2010, 48(1): 132-144.
[18]Oberlin A. Pyrocarbons[J]. Carbon, 2002, 40(1): 7-24.
[19]Deutschmann O, Tischer S, Correa C, et al. DETCHEM Software Package, 2.1 ed. www.detchem.com, Karlsruhe, 2007.
[20]Frenklach M, Wang H. Detailed surface and gas-phase chemical kinetics of diamond deposition[J]. Physical Review B, 1991, 43(2): 1520-1545.
[21]Wauters S, Marin G B. Kinetic modeling of coke formation during stream cracking[J]. Ind Eng Chem Res, 2002, 41(10): 2379-2391.
[22]Celnik M, Raj A, West R, et al. Aromatic site description of soot particles[J]. Combustion and Flame, 2008, 155(1-2): 161-180.
[23]Okkerse M, Croon M de, Kleijn C R, et al. A surface and a gas-phase mechanism for the description of growth on the diamond 100 surface in an oxy-acetylene torch reactor[J]. Journal of Applied Physics, 1998, 84(11): 6387-6398.
[24]Marinov N M, Pitz W J, Westbrook C K, et al. Aromatic and polycyclic aromatic hydrocarbon formation in a laminar premixed n-butane flame[J]. Combust Flame, 1998, 114(1-2): 192-213.
[25]Violi A, Truong T N, Sarofim A F. Kinetics of hydrogen abstraction reactions from polycyclic aromatic hydrocarbons by H atoms[J]. J Phys Chem A, 2004, 108(22): 4846-4852.
[26]Hemelsoet K, Van Speybroeck V, Moran D. Thermochemistry and kinetics of hydrogen abstraction by methyl radical from polycyclic aromatic hydrocarbons[J]. J Phys Chem A, 2006, 110(50): 13624-13631.
[27]Richter H, Benish T G, Mazyar O A, et al. Formation of polycyclic aromatic hydrocarbons and their radicals in a nearly sooting premixed benzene flame[J]. Proc Combust Inst, 2000, 28(2): 2609-2618.
[28]Ziegler I, Fournet R, Marquaire P M. Influence of surface on chemical kinetic of pyrocarbon deposition obtained by propane pyrolysis[J]. J Anal Appl Pyrolysis, 2005, 73(1): 107-115.
Modeling of carbon deposition from propane in chemical vapor infiltration
TANG Zhe-peng1,XU Wei1,LI Ai-jun1,ZHANG Zhong-wei2,BAI Rui-cheng1,WANG Jun-shan2,REN MU-su1
(1.SchoolofMaterialsScienceandEngineering,ShanghaiUniversity,Shanghai200072,China;2.NationalKeyLaboratoryofAdvancedFunctionalCompositeMaterials,AerospaceResearchInstituteofMaterialsandProcessingTechnology,Beijing100076,China)
A reaction model including global reactions and 1 074 elementary reactions of 285 species in both the gas phase and the solid surface was used to numerically simulate the gas phase composition and the deposition kinetics of pyrocarbon on carbon fibers in chemical vapor infiltration using propane. The global reactions were simplified to be the direct dehydrogenation reactions from the hydrocarbon species in the gas phase while the elementary reactions included 66 surface species and 250 elementary steps. Simulated results were compared with experiments performed in a perfectly stirred reactor at 2.6 kPa from 1 173 to 1 323 K for 0.5 to 4 s, using propane as the carbon precursor and nitrogen as the diluting gas. Excellent agreement between the simulated and the experimental results are found for both the gas phase compositions and deposition kinetics at various conditions. The simulation indicates that the pyrocarbon precursors are mostly small unsaturated species (acetylene and ethylene) and methyl radicals, and the deposition kinetics could be quantitatively described by the deposition of these species.
Modeling; Pyrocarbon; Surface kinetics; Propane; Chemical vapor infiltration
Doctoral Fund of Ministry of Education (20113108120019); Shanghai Talent Development Fund (2011028); Aviation Fund (2013ZFS6001); Shanghai Committee of Science and Technology(13521101202).
LI Ai-jun, Professor. E-mail: aijun.li@shu.edu.cn
introduction: TANG Zhe-peng, Ph. D Candidate. E-mail: tangzhepong@shu.edu.cn
1007-8827(2016)01-0077-10
TB332
A
2015-11-20;
2016-01-02
教育部博士点基金(20113108120019);上海人才发展资金(2011028);航空基金(2013ZFS6001);上海市科委基金(13521101202).
李爱军, 东方学者, 教授. E-mail: aijun.li@shu.edu.cn
汤哲鹏, 博士研究生. E-mail: tangzhepong@shu.edu.cn
