基于生物刺激源的响应性聚合物及其可控自组装
2016-10-26马明煊复旦大学高分子科学系聚合物分子工程国家重点实验室上海200433
张 建, 马明煊, 闫 强(复旦大学高分子科学系,聚合物分子工程国家重点实验室,上海200433)
特约综述
基于生物刺激源的响应性聚合物及其可控自组装
张 建, 马明煊, 闫 强
(复旦大学高分子科学系,聚合物分子工程国家重点实验室,上海200433)
基于生物刺激源的响应性大分子自组装体系对智能聚合物领域的发展具有重要的意义。本文回顾并综述了近年来在该领域内针对生物大分子及各种小分子作为刺激源的大分子自组装体系及其研究现状。依据不同类型的刺激源进行分类,论述了近年来的研究进展,并合理展望了该领域未来的发展前景。
生物刺激源;响应性大分子;可控自组装
刺激响应性聚合物(stimuli-responsive polymer)是智能大分子的一种类型,其受到一定的因素(输入信号)扰动时能够发生结构与性质的变化,同时显示功能变化(输出信号)[1]。这一类高分子作为智能大分子的主体,具有优良的物理与化学性能,并且在纳米技术[2]、功能材料[3]、生物成像与传感[4-5]等诸多领域拥有巨大的应用前景,在纳米科学、材料科学与仿生科学等不同交叉学科都发挥着突出作用,引领着功能高分子材料发展的方向[6-7],因此受到人们越来越多的关注与研究,成为现代高分子科学的一个重要研究领域。
刺激响应性聚合物能够在生理环境下自组装形成特定形状(如球状胶束或囊泡等)的智能纳米载体(smart nanocarriers),当在内部负载药物分子后,基于大分子本身对病理细胞的高渗透/滞留效应(EPR)与刺激响应性解离的特性,能够有效地实现对负载分子可控释放的功能,同时降低对正常细胞的毒副作用[8]。因此,刺激响应性聚合物在纳米药物、临床诊断、靶向治疗、医学成像等生物医学前沿蕴藏着巨大应用价值。
近些年来国内外对于刺激响应性聚合物的研究与应用一直都着眼于不同类型的刺激源开发中[9]。针对响应型聚合物组装体系的开发与利用取得了瞩目的进展,依托于传统的外源性物理或化学刺激源已形成了较成熟的聚合物组装体系,比如,Armes课题组[10]以聚N,N-二乙基甲基丙烯酸乙二胺酯为质子敏感性链段制备了p H响应型聚合物胶束。之后,国内外针对传统化学或物理刺激条件的开发,包括p H、氧化还原剂、离子、温度、磁场、电场、光辐射等取得了极大的进展,基于传统刺激方式而制备的响应性聚合物组装体系也已日渐完备[11-25]。
但需要指出的是,这一类聚合物最值得人们关注的一点就是某些刺激响应类型与生物体系中的生物信号应激行为类似,这就为揭示生物活性机制以及指引仿生材料的发展提供了无限的可能性[26]。近年来,许多刺激响应性聚合物的新兴以及前瞻性的应用要求其可以适应人体的生理条件(多为细胞内利用),但是传统的刺激响应模式并不能满足某些特定的需求,比如,包括基于p H、氧化还原剂在内的大多数刺激模式,需要相应化学制剂的连续加入,这些物质在体内经过多次刺激-应答循环后不断累积,不易被清除或被细胞分解,且被证实会引起细胞毒性[27]。除此之外,电场能、磁场能、机械力或紫外光辐射能等[28]刺激模式会在一定程度上造成基因突变或细胞永久性损伤。另外,在纳米载体领域,响应性聚合物纳米载体的作用方式是利用正常细胞与病理细胞间生理微环境的差异性进行靶向输运与控制释放。但大部分疾病已被证明胞内环境差异有限,传统刺激方式与聚合物结构之间的选择性与特异性不足,聚合物组装体进入生物体内后未达到病灶处即被正常细胞的生理环境所解离,难以实现药物的有效生物分布[29]。因此,为了消除在刺激响应过程中副产物的产生并满足可以在人体内进行应用的要求,开发新型的刺激模式就变得迫在眉睫。
由此,人们设想是否可以直接采用细胞内存在的各种内源性生物信号作为刺激源(生物刺激源)构建聚合物组装体系,如细胞信使分子、激素、代谢产物、维生素、脂类和酶等,这将是刺激响应性聚合物领域崭新且极有意义的发展方向。这是因为:(1)生物信号分子属于细胞天然代谢或分泌物,无生理毒性,可有效避免细胞损伤,其内源特质不存在化学污染物累积问题;(2)疾病产生的病理本质是由于分子层次上某种特定生物信号分子或特定蛋白的代谢异常所引起,这势必导致正常细胞与病理细胞之间该生物分子的显著性浓度差异,利用该特异性差别,可以呈几何级数形式放大对该种生物分子的特异性响应能力;(3)生物信号分子范围广泛,本身隶属于生物化学、分子生物学、细胞生物学等学科的研究对象,利用其作为刺激源在响应性聚合物中应用,势必进一步推动聚合物科学与上述众多学科之间的交叉与联系,从而拓延新生的科学知识。
生物刺激源根据其分子类型主要划分为两类:
(1)生物大分子型,包括酶(enzyme)、DNA等;(2)小分子生物信号,包括神经信号转导分子(cell signaling)、代谢物(metabolite)、生物活性分子(bioactivator)、反应活性物种(reactive species)、激素(hormone)、维生素(vitamin)等(如图1所示)。接下来,本文将根据构建响应性大分子组装体系的生物刺激源种类展开叙述。
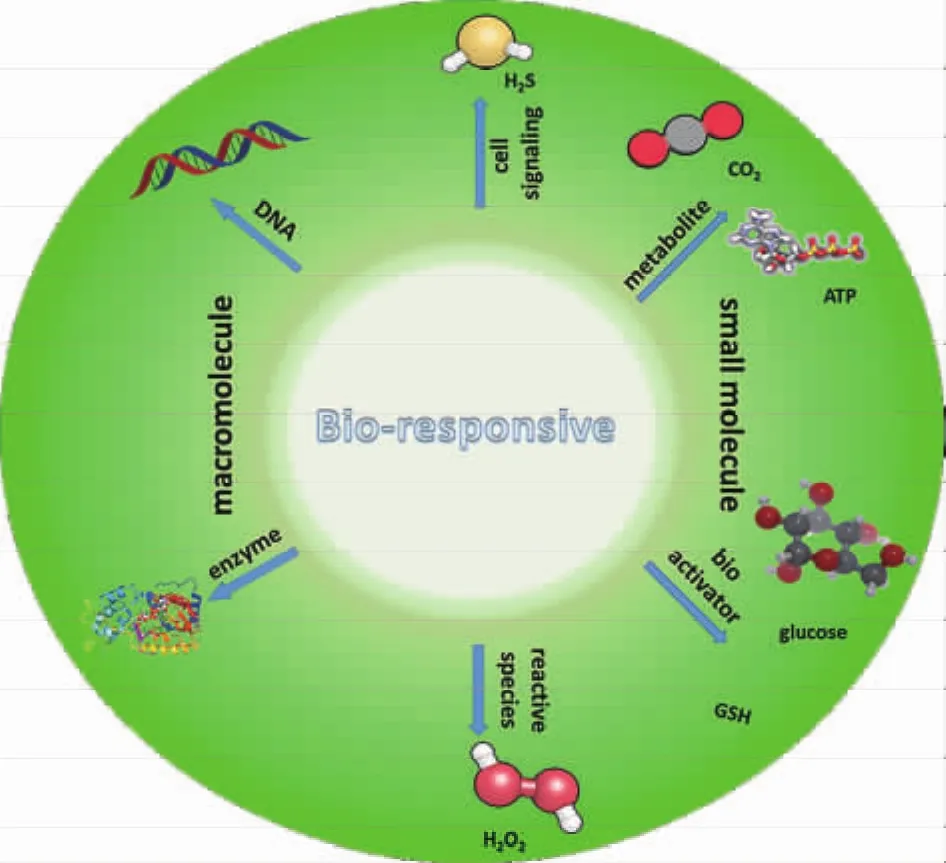
图1 生物刺激源响应的大分子可控自组装Fig.1 Controllable self-assembly of biological stimuli-responsive polymers
1 生物大分子型
1.1酶
通常病态细胞伴随着某种酶的过度表达,同时由于良好的生物兼容性以及高度的选择性[30],酶响应性的聚合物组装体受到人们广泛的青睐。比如,在多种类型的肿瘤细胞中某些酶的浓度高于正常水平,那么由此就可以构建基于酶响应的载药体系用于抗肿瘤药物的缓释与癌症的治疗[31]。
近5年来国内外在该方向上的研究进展主要集中于对大分子蛋白的刺激方式开发上。比如:Thayumanavan等[32]在近期选用功能化基团修饰的两亲性多肽作为模板,以苯磺酰胺-牛碳酸酐酶(bCA-Ⅱ)之间的相互作用为驱动力,实现了酶响应性的聚合物解组装以及模型药物的释放,如图2所示。作者指出,尽管响应性分子组装体已经被用于药物的输送与诊断领域,但是针对蛋白质活性响应的相关体系还非常有限,而异常的蛋白质活性在所有的遗传性疾病中都很普遍。虽然针对酶活性变化的组装体系已经取得了重要的进展,但针对非酶蛋白质的响应性组装体却鲜有报道。多肽类聚合物由于其良好的生物兼容性、生物可降解性与高保真响应解组装等特性,将会在该领域大放异彩。
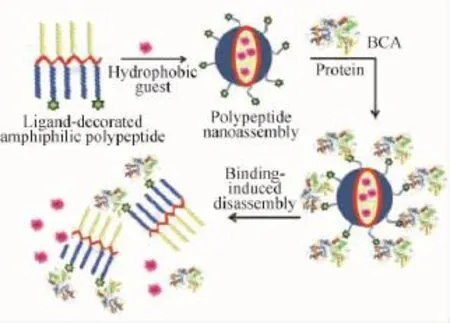
图2 基于蛋白质-配体结合机制的多肽类聚合物解组装体系[32]Fig.2 Polypeptide assemblies based on protein-ligand interactions[32]
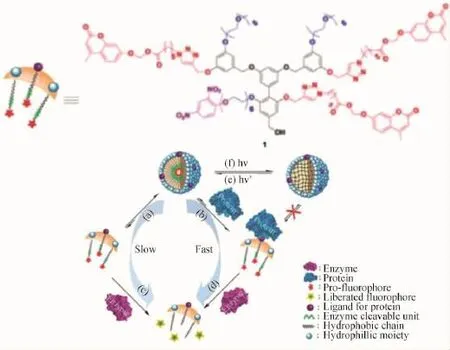
图3 蛋白与脂酶双控的超分子解组装体系[33]Fig.3 Protein and lipase dual-triggered supramolecular disassembly system[33]
Thayumanavan等还设计了一种两亲性的树枝状聚醚(如图3所示),并实现了蛋白与脂酶双控的超分子解组装体系[33]。其中,疏水的香豆素酯位于两亲性类胶束组装体内部而避免了与脂酶的接触,但是由于组装体亲水表面的二硝基苯单元可以与anti-DNP免疫球蛋白(IgG)结合使得两亲性聚醚单分子与聚集体之间的平衡被打破(平衡移向单分子),从而导致组装体的崩塌;此时疏水端的香豆素酯单元暴露并与脂酶作用而使香豆素的荧光发射恢复。除此之外,该团队通过将这一类聚醚引入苯磺酰胺与生物素末端官能团,还先后实现了对生物蛋白碳酸酐酶与亲和素刺激响应的解组装体系。该课题组还首次开发了一类基于蛋白质-配体结合机制的聚合物解组装体系,如图4所示。首先,将一种蛋白质配体(biotin)修饰到两亲性树枝状聚醚的亲水侧,在水中该树状聚醚自组装形成类胶束组装体并包裹疏水性客体分子。当加入一种抗生物素蛋白(extravidin)后,由于biotin与extravidin之间的结合作用,胶束表面的配体被亲水性的蛋白所取代,导致树状分子亲疏水平衡的剧烈变化,从而使得其组装体结构解离,其包裹的客体分子释放[34]。
Khan等[35]将亲水性的聚乙二醇(PEG)链段和疏水性的聚苯乙烯(PS)链段通过偶氮相连接,由此构建的两亲性嵌段聚合物可以在水相中自组装形成胶束结构,如图5所示。在偶氮还原酶与辅酶NADPH的作用下,聚合物中的偶氮连接处断裂,形成独立的PEG链段与PS链段,由此胶束组装体崩塌,新生成的PEG链段溶于水中,PS链段形成沉淀。这一结果表明偶氮苯可以作为一种有价值的非天然功能化单元用于酶响应性聚合物组装体的构建。而由于偶氮还原酶的敏感性,这一类材料的潜在应用价值体现在靶向结肠的特异性药物输运体系等方面。
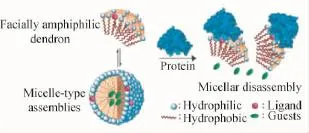
图4 基于蛋白质-配体结合机制的聚合物解组装体系[34]Fig.4 Polymer disassembly system based on protein-ligand interactions[34]
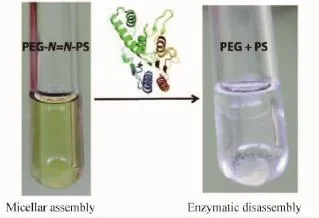
图5 应答偶氮还原酶的聚合物组装体[35]Fig.5 Azoreductase-sensitive polymer assemblies[35]
于此同时,张希课题组[36]通过三磷酸腺苷(ATP)-离聚物复合物开发了磷酸脂酶响应型的聚合物胶束,其机制如图6所示。
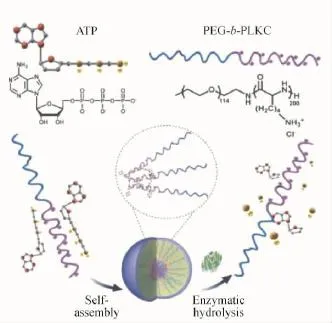
图6 基于ATP-离聚物复合物的磷酸脂酶响应型聚合物胶束[36]Fig.6 Phospholipase-responsive ATP-polymer ionomer complex[36]
众所周知,ATP在生理条件下包含有一个疏水的腺嘌呤单元,并且其膦酸苷键可以被磷酸酶水解而由多负电荷的分子转变为中性的腺嘌呤。由此,作者利用带有正电荷链段的两嵌段聚合物(PEG-b-PLKC)(PLKC为聚赖氨酸)与ATP之间的静电相互作用,构建以腺嘌呤为疏水核的ATP-离聚物复合物胶束。随着磷酸酶的加入,ATP开始水解而使之前的复合物解离,组装胶束也随之崩塌。
1.2DNA
DNA具有良好的生物兼容性与可程序化控制性,同时兼具易于制备、便于修饰的特点,因此成为理想的结构构筑材料,并被应用于DNA纳米技术领域[37]。人们已经将有机与无机合成分子修饰的DNA用于构筑超分子DNA组装体[38],从而使得DNA纳米结构的功能性更为丰富。利用这些三维结构,人们已经可以实现最小DNA的柱状结构组装,精确调控其纳米管的相关参数,以及在其内部营造疏水环境等[39]。
Sleiman等[40]使用DNA短链构建了类脂质分子修饰的DNA“立方笼”,这些脂质可以通过分子内相互作用在DNA“立方笼”内相互“握手”(如图7所示),从而形成稳定的胶束结构并将药物模型(尼罗红)包裹,在特定核酸序列的情形下,“立方笼”内成核的单元通过序列取代的方式被“擦除”,药物模型得以释放。包括肿瘤细胞在内的多种病变细胞存在某种基因的过度表达,所以在以后的应用中,DNA“立方笼”可将药物输运至病变细胞的环境中,从而触发药物释放。随后,该课题组[41]设计了一种动态的DNA“立方笼”,此“立方笼”可以在特定核酸序列(存在于前列腺癌细胞中)识别的基础上选择性地打开,并形成二维结构,如图8所示。这一“立方笼”架构对核酸酶表现出良好的稳定性,甚至在耐药性的细胞株中都表现出优良的细胞吸收行为。作者用疏水与亲水的树状DNA链段修饰该“立方笼”后发现,在维持其靶向序列响应能力与其在生物环境中稳定性的前提下,“立方笼”被细胞吸收的行为大大改变。作者指出,这项工作为特异性响应(针对病变细胞中过表达的核酸序列)的DNA“立方笼”的创建提供了坚实的基础,这一研究可能被用于输运多种靶向治疗的核酸药物。
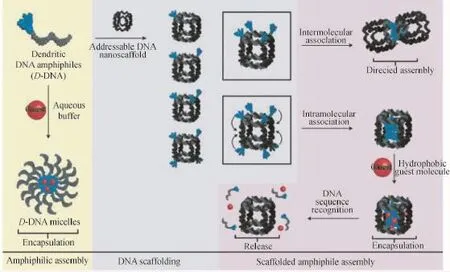
图7 DNA响应性纳米“立方笼”载运体模型[40]Fig.7 DNA-responsive“nanocube”carriers[40]
2 小分子生物信号
与生物大分子相比,小分子类生物信号在细胞内的分布更加广泛,对相关生物体机能的调控更加敏感,细胞穿透性更好,在生物体与细胞内的浓度也远高于生物大分子,因此从作为生物刺激源的角度来说,开发基于小分子类生物信号的刺激响应性聚合物具有更大的应用价值。但是,生物体以及细胞中存在多种结构或者性质类似的生物活性小分子(以H2S为例,细胞中共存有半胱氨酸(Cys)、高半胱氨酸(Hcy)、谷胱甘肽(GSH)等多种性质相似的含硫物种[42]),因此合理设计对特定生物信号具有特异响应性的聚合物结构成为解决上述问题的关键所在。
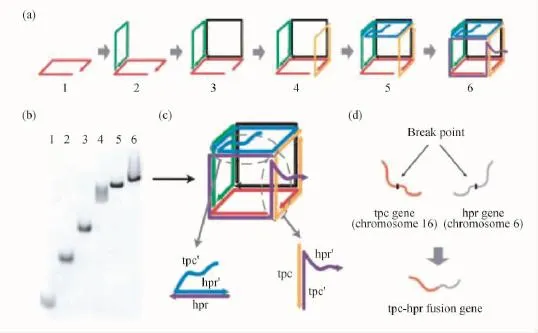
图8 具有特异响应性的动态DNA“立方笼”[41]Fig.8 Specific-responsive dynamic DNA“nanocube”[41]
2.1神经信号转导分子
2.1.1硫化氢 众所周知,H2S是一种大气污染物,但它同时也是一种非常重要的神经调质与细胞信号分子。在细胞中,H2S由L-半胱氨酸经过胱硫醚γ-裂解酶分解生成[43]。最新的研究表明,H2S在调节血管扩张,抗发炎以及诱导细胞凋亡等方面有着重要的作用[44]。H2S的代谢紊乱与包括心血管病以及神经衰退型疾病在内的多种疾病直接相关。考虑到H2S在细胞中的重要性,我们课题组最近开发了一种利用H2S作为刺激源的聚合物组装-解组装体系,并成功地实现了相应靶向药物(肾上腺素)的输运[45]。
我们设计并合成了一种新型的含有双取代苄基酯(Az MB)类结构的两亲性聚合物(如图9所示),该聚合物可以在水相中自发组装形成囊泡结构,并且功能化响应基团赋予这些囊泡对于生物信号分子H2S独特的灵敏性。H2S可以通过Az MB特定位点的断裂促发聚合物囊泡可控地解组装并由此改变聚合物的两亲性,从而导致聚合物囊泡的可控解组装行为。由此,我们认为如果将可以产生H2S的蛋白质修饰在囊泡膜上,那么从某种程度上来说,该聚合物的响应范围就可以从简单的H2S扩展到较为复杂的生物分子。从药物载运的角度来说,这种内源性刺激响应模式的纳米载体有望应用于细胞内靶向释放与治疗,与此同时这一类聚合物模型也为构建生物刺激响应的纳米胶囊提供了新的思路与方向。
2.1.2一氧化氮 一氧化氮(NO)虽然对人体健康具有潜在的危害,但它可以作为内皮细胞舒血管因子(EDRF)以及生物信号分子,在细胞水平上发挥重要的作用[46]。NO是一种在活体组织中生物合成的、半衰期较短(仅为几秒)的气态自由基物种,它在生物体的心血管、神经与免疫系统中承载信使分子的角色[47],并可以影响其响应的生理活性[48]。人体中NO浓度异常可导致心血管障碍,胃肠痛,神经变性以及高血压等多种疾病[49]。由此,Davis等[50]开发了一类NO刺激响应的聚合物(P(NIPAM-co-NAPMA)与P(NIPAM-co-APUEMA)),其响应机制在于聚合物中的邻苯二胺单元与NO之间的反应:对于聚合物P(NIPAM-co-NAPMA),相应的酰胺键取代的苯并三唑衍生物中间体可以自发地水解生成苯并三唑;而对于P(NIPAM-co-APUEMA),脲功能化的苯并三唑衍生物却可以稳定存在。作者同时证实,可以利用NO的刺激作用调节两种温敏性聚合物的低临界溶解温度(LCST)值(显著升高与降低)。更重要的是,依靠NO(极低浓度)触发的疏水基元的形成,可以精确调控聚合物P(NIPAM-co-APUEMA)的自组装(胶束)过程。这一研究为NO刺激响应的药物释放奠定了非常重要的理论基础,如图10所示。
2.2代谢物
2.2.1二氧化碳 由于温室气体效应CO2受到人们普遍重视的同时,有越来越多的研究表明,它作为新陈代谢的一种主要产物,已经被证明与包括血碳酸过多症在内的某些生理疾病有关[51],而对生物体动脉与静脉CO2水平的监测也会为生理状态的诊断提供重要的依据[52-53]。另外,CO2具有良好的生物兼容性与生物膜穿透性[54],因此它可以作为一种刺激源应用于人体生理环境。[]
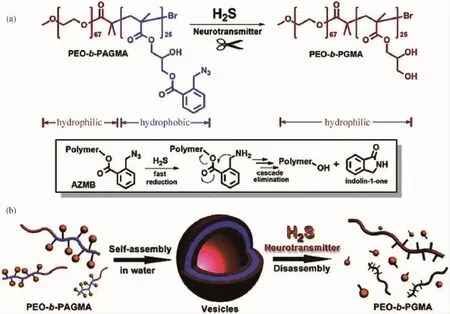
图9 基于H2S刺激响应的聚合物囊泡[45]Fig.9 H2S-responsive polymersomes[45]
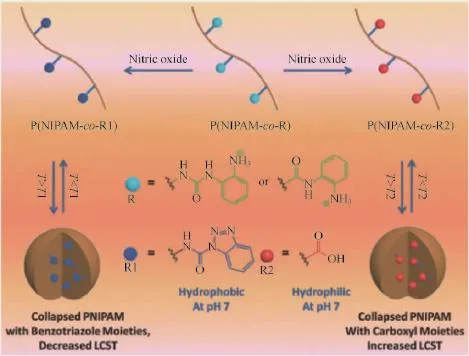
图10 基于NO刺激响应的温敏性聚合物50Fig.10 NO-responsive thermally sensitive polymers[50]
CO2可以选择性地与某些功能基团(如叔胺基、脒基、胍基等)反应而使之转变成亲水性物种,同时惰性气体(氩气或氮气)又可以很容易地实现上述反应的逆过程,这意味着该可逆过程可以在没有任何化学制剂累积的情况下实现多次“开-关”循环[55]。因此,对于聚合物组装体系而言,CO2是一种真正意义上的温和的“绿色”刺激源。本课题组自2010年开始,一直致力于以CO2为刺激源的聚合物组装-解组装体系研究,利用聚合物对CO2的类“呼吸”行为,成功开发了基于CO2刺激响应的一系列功能化应用,包括仿生纳米容器、蛋白分离、仿生细胞等。
2010年[56]我们利用一种含有脒功能基团的两亲性嵌段共聚物(PEO-b-PAD)(PAD为聚十二烷基脒基丙烯酰胺),成功实现了其囊泡状组装体在水溶液中对CO2的刺激响应,使其可以进行仿生“呼吸”运动(如图11所示)。首先,PEO-b-PAD自组装形成尺寸为110 nm,膜厚22 nm的囊泡状组装体;随着CO2对脒基团质子化程度的提高,囊泡的体积增大到205 nm;当用Ar刺激上述体系时,体积增大的囊泡可逆地缩减到原来的尺寸,该过程类似于肺泡细胞的“呼吸”式运动。
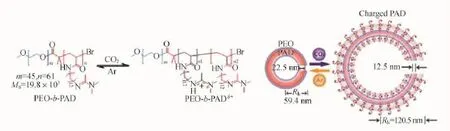
图11 CO2响应性嵌段共聚物PEO-b-PAD组装体的仿生“呼吸”运动[56]Fig.11 CO2-responsive PEO-b-PAD copolymer for mimicking“breathing”motion[56]
随后,我们证实利用该聚合物对CO2的刺激响应性,在通过调节CO2对聚合物中的脒嵌段的质子化程度而改变其囊泡组装体大小的同时,也可以实现对囊泡膜通透性的调控[57]。利用这一特性,我们可以实现对某些组分的逐级释放与客体分子的选择性分离。此外,该囊泡可以像细胞一样区分不同类型的酶催化反应。我们期望这种可控的囊泡结构可以帮助人们更好地模拟细胞膜结构并进行更深层次的应用。
高尔基体是一种重要的细胞器,主要功能在于对蛋白质的修饰与传递。在该过程中高尔基体可以展现出多种不同的自组装形貌(从细管经过囊泡到柱状体与球等)以实现上述不同的功能。最近我们利用一种对CO2刺激响应的三嵌段共聚物PEO-b-PAD-b-PS,实现了对高尔基体组装形貌的模拟过程[58](如图12所示)。
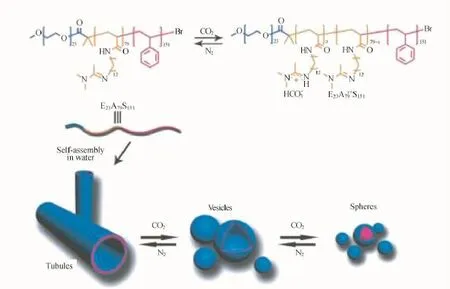
图12 CO2响应性三嵌段共聚物PEO-b-PAD-b-PS对高尔基体组装变形的模拟[58]Fig.12 CO2-responsive deformation of PEO-b-PAD-b-PS triblock copolymer assemblies Golgi apparatus motion[58]
具体来说,该聚合物以疏水性的PS为内核,对具有CO2刺激响应性的PAD为中间层,外层为亲水性的PEO。在水相中,聚合物可以自组装形成一种刚性的管状形貌(其轴向长度可以达到微米尺度,直径为320 nm到680 nm不等),将CO2通入此溶液中15 min,该管状结构完全消失,取而代之的是平均尺寸为410 nm的亚微米囊泡。当通入气体25 min时,溶液中的组装形貌从囊泡经由“串珠状”柱状体转变为球状胶束,该过程刚好与细胞中高尔基体的形貌演变类似。上述聚合物组装体形貌转变的机制在于:聚合物中间链段PAD受CO2不断刺激而质子化程度逐渐增加,从而导致聚合物整体亲水链段比重的持续升高。在CO2刺激之前,由于聚合物链之间较弱的相互作用以及紧密的链排列,聚合物组装成曲率最低的管状结构;在CO2刺激下,PAD链段中的脒基团质子化成为脒盐,外层链之间的静电排斥作用力增强,迫使管状聚集体为了最小化其表面自由能而减小体积,形成较高曲率的囊泡结构;进一步的CO2刺激将会使得静电排斥力进一步增加,从而使组装体进一步升高曲率而形成柱状体与球状胶束。因此,通过调节CO2浓度而调控嵌段共聚物亲疏水链段比例,可以准确地控制由该三嵌段聚合物形成的自组装聚集体的界面曲率以及形貌。
作为一种生物体的固有特性,在生理条件刺激下细胞器的形变对相应生物功能的执行以及维持细胞活性至关重要。比如,一些细胞器展现出自发式的运动,细胞内的脂滴细胞器可以经过某些活性的生物分子自调节它们的球状尺寸,以储存或者释放其内部的营养素;微丝经历有丝分裂的过程中可以改变其形状(从卷曲型转变为伸直的纤维)以帮助细胞的运动;溶酶体具有内吞作用,其膜可以通过凹入与裁剪避免有害固体颗粒的摄入(如图13所示)。
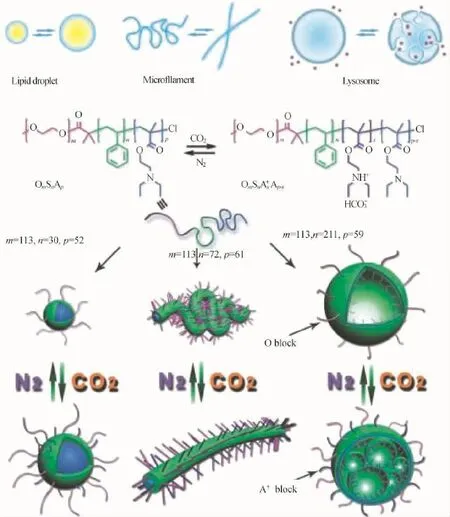
图13 CO2响应性三嵌段共聚物(OSA)对细胞器变形的模拟[59]Fig.13 CO2-responsive triblock copolymer OSA for mimicking the deformation of organelles[59]
我们通过调换CO2响应链段与疏水性PS链段的位置,成功地实现了对细胞器形变过程的模拟[59]。该三嵌段共聚物(OSA)依次由外层亲水性的PEO链段(O),中间疏水性的PS链段(S)以及对CO2具有刺激响应性的聚N,N-二乙基甲基丙烯酸氨乙酯(PDEAEMA)链段(A)构成。保持相同长度的O链段以及相似长度的A链段,通过改变S链段的程度,OSA共聚物可以自组装形成3种初始的纳米结构,分别为球状胶束,蠕虫状胶束与巨型囊泡(此时A链段均位于核内部)。对于含有最短S链段的聚合物O113S30A52,通过CO2的连续加入,其球状胶束组装体尺寸可以实现从24 nm到45 nm的连续增大,这与脂滴的自调节“呼吸”特性类似;对于含有较长S链段的聚合物O113S72A52,其在水相中可组装形成高长度-直径比例的卷曲与折叠状的纳米纤维结构,随着CO2的连续加入,其初始的卷曲纤维转变成平直纳米线,这刚好与微丝的弹性伸缩运动类似;对于含有最长S链段的聚合物O113S72A52,在水相中可以形成巨型囊泡(600 nm)的组装结构,随着CO2的连续加入,巨型囊泡被微区化为较小尺寸(40~180 nm)的由连续膜分离的不规则液泡,该过程从一定程度上来说,与溶酶体的内吞行为类似。上述基于OSA三嵌段共聚物的各种组装体形变的关键在于CO2响应片段PDEAEMA(初始具有疏水性)位于疏水核的内部并被S链段限制,当A链段受CO2质子化后电荷化与亲水性逐步增加,虽然不能自由溶解,但它可以吸收更多的水。这项研究表明,外部链段的相互排斥与内部链段的受限水合的协同作用,是一种有效的控制CO2刺激响应的三嵌段聚合物组装变形的策略,而且这一类CO2调控的聚合物模型为仿生提供了全新的研究思路。
最近,黄飞鹤等[60]以二乙胺基修饰的柱芳烃(Pillar[5]arene)与十二烷基磺酸钠(SDS)构建了CO2刺激响应的超分子囊泡,如图14所示。将CO2通入体系中,柱芳烃的二乙胺基质子化而带正电荷,这刚好与SDS亲水的负电荷部分相互吸引,同时其疏水的空腔刚好接纳SDS的长链烷基,两者协同作用形成的双层分子囊泡组装体在N2的作用下解组装而使包裹的药物释放。作者指出,这种新型的CO2刺激响应的自组装体系在药物载运与传感方面具有潜在的应用价值,同时对人们更好地理解生物过程大有裨益。
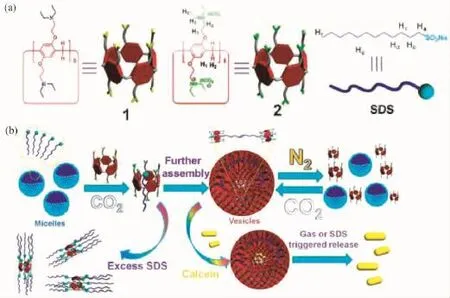
图14 CO2刺激响应的超分子囊泡[60]Fig.14 CO2-responsive supramolecular vesicles[60]
2.2.2ATP ATP作为一种细胞内的能量货币与细胞内至关重要的生物信号分子,在多种细胞活性中表现出不可替代的作用。ATP已经被作为刺激信号用于调控纳米载体的解组装过程。比如,Aida等[61]通过一种用螺吡喃修饰的桶状分子伴侣蛋白GroEL与镁离子的配位作用,构筑了一类管状纳米结构(如图15所示)。受细胞内ATP水解所提供的能量影响,分子伴侣蛋白GroEL发生形变,进而导致管状纳米结构的解组装而使客体分子释放。
随后,该课题组利用ATP与胰蛋白酶Trp对含有功能化胍盐与硼酸单元的分子胶水的竞争黏附能力,实现了经由ATP对胰蛋白酶活性的调控,并取得了初步的研究成果[62],如图16所示。
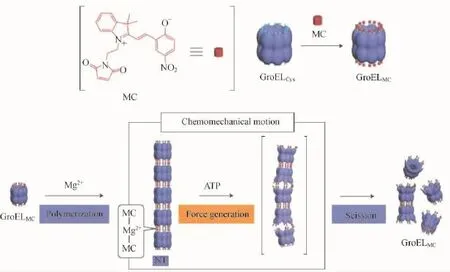
图15 基于ATP响应的GroEL蛋白生物大分子纳米管形变[61]Fig.15 ATP-responsive GroEL protein tubules and their shape transformation[61]
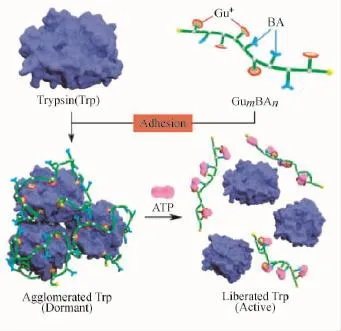
图16 基于ATP竞争黏附的聚合物复合体对Trp的活性控制[62]Fig.16 ATP competitive-based polymer complexes for controlling Trp activity[62]
Gu等[63]利用ATP响应性的适配体DNA与其互补的链段作为载体包裹阿霉素(DOX),实现了在ATP刺激下药物的释放,如图17所示。
近期我们首次利用ATP作为刺激源,成功调控了其对合成聚合物的组装行为[64]。受天然ATP结合蛋白结构的启发,我们设计了仿生ATP识别单元(如图18所示),利用腺嘌呤-环糊精的主客体相互作用与三磷酸盐-胍的氢键的协同效应,实现了对ATP分子的特异响应性,并抑制了胞内ATP相似物(如二磷酸腺苷ADP,单磷酸腺苷AMP,三磷酸胞苷CTP,三磷酸鸟苷GTP等)的干扰。通过对ATP的捕获,聚合物可以与之形成杂交复合物,我们可以通过调控ATP的刺激水平而调节杂交复合物中亲疏水链段比例的新平衡,从而准确控制预期的自组装结构的形貌、维度与变形行为。我们希望这种生物刺激响应的聚合物模型可以用于构建智能仿生组装,模拟细胞与细胞器的运动。
黄飞鹤等[65]近期制备了基于柱芳烃的大分子纳米胶束,通过对ATP水解的抑制,有效地提高了抗癌药物的药效(如图19所示)。其机制在于:末端修饰叶酸(FA)的两嵌段共聚物聚乙二醇-b-聚丙烯酸(FAPEG-b-PAA)与正离子型水溶性柱芳烃(WP6)形成聚离子型胶束后将WP6载入细胞,WP6可以对ATP特异性结合(化学计量比1∶1)而生成稳定的WP6复合物,从而导致ATP水解受阻,使癌细胞中的ATP依赖的多耐药性(MDR)受到抑制,细胞内药物蓄积增加。这项研究表明,人们可以通过利用超分子化学设计理想的载运体用于克服癌症治疗过程中的多耐药性,这也为发展新型的诊疗试剂提供了新的思路。

图17 基于ATP响应的适配体DNA载药模型[63]Fig.17 ATP-responsive DNA aptamers and drug delivery[63]
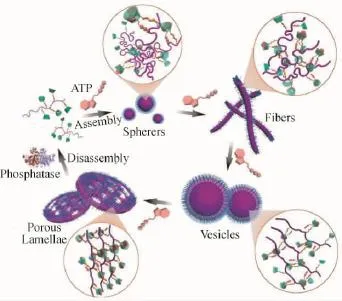
图18 ATP分子特异性驱动的大分子组装体形态转变行为[64]Fig.18 ATP specific-responsive polymer assemblies deformation[64]

图19 WP6抑制ATP水解[65]Fig.19 WP6-based polymer micelles for ATP hydrolysis inhibition[65]
2.3 生物活性分子
2.3.1葡萄糖 糖尿病已经成为一种影响全球公共卫生事业的疾病,其主要表现为血液中葡萄糖浓度的累积,因此,人们发展了一系列葡萄糖响应的聚合物载药(胰岛素)体系以用于糖尿病的治疗。在多种葡萄糖响应体系中,最受关注的就是以硼酸及其衍生物为主体的一类聚合物,其原因在于:(1)硼酸及其衍生物便于引入到不同的设计体系中,并且要比基于蛋白质(葡萄糖氧化酶与伴刀豆球蛋白A)的大分子体系稳定性更高[66];(2)通过不同分子的加入,相应的聚合物组装体更加便于修饰与功能化[67-69],并使得预装载的蛋白质药物免受外部环境的破坏[70]。比如,Li等[71]通过可逆加减-断裂链转移(RAFT)聚合分别得到含有苯硼酸与吡喃葡萄糖链段的两亲性嵌段与随机共聚物,这两种共聚物均可自组装形成稳定的窄分布球形纳米粒子并作为药物载体包裹胰岛素(如图20所示)。作者发现与嵌段共聚物纳米粒子相比,随机共聚物纳米粒子在葡萄糖刺激下,胰岛素的释放具有更快的速率与更多的累积量,而糖链段的引入使得苯硼酸单元的生物兼容性提高,确保共聚物可以在生物医药领域得以应用。
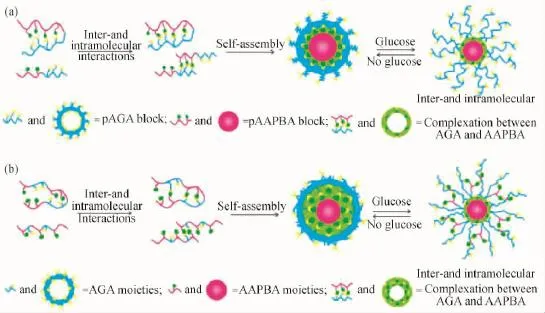
图20 基于葡萄糖刺激响应的聚合物载药体系[71]Fig.20 Glucose-responsive polymer nanocarriers for drug release[71]
Kim等[72]以PEO为亲水链段,含苯硼酸衍生物的链段为疏水链段,构建了可以对单糖(包括葡萄糖)在中性p H条件下进行刺激响应的聚合物解组装体系。如图21所示,由于硼酸单元与单糖中邻二醇之间特异性的结合作用,聚合物组装体解组装,封装在聚合物囊泡中的胰岛素得以释放。上述体系有望在葡萄糖相关的疾病(比如糖尿病)的治疗中作为传感探针与载药体系得到有效的利用。
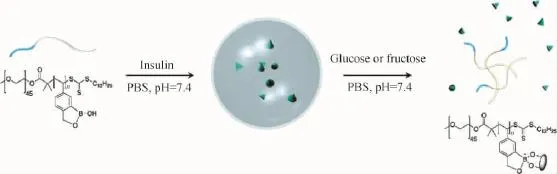
图21 基于单糖刺激响应的聚合物载药体系[72]Fig.21 Monosaccharide-responsive polymer nanocarriers and drug release[72]
2.3.2生物硫醇 有研究表明谷胱甘肽(GSH)与氧化谷胱甘肽(GSSG)是动物细胞中最为丰富的氧化还原对。在细胞质与核酸中GSH的浓度在谷胱甘肽还原酶的作用下达到10 mmol/L,而在细胞外其浓度为2~20 mmol/L[73]。小鼠活体研究表明,肿瘤组织中GSH的浓度至少是正常组织的4倍[74],因此,利用该差异构建GSH刺激响应的载药系统就变得十分重要。Zhong等[75]以二硫键修饰的PEG为亲水链段,以含有p H响应的缩醛单元的季戊四醇碳酸酯为疏水链段,开发了一类氧化还原与p H双响应的可生物降解的聚乙二醇-聚(2,4,6-三甲氧基苯亚基季戊四醇碳酸酯(PEG-SS-PTMBPEC)载药体系(如22图所示)。由PEG-SS-PTMBPEC形成的胶束粒子的平均尺寸为140 nm,该胶束可以包覆DOX,包覆的质量分数可以达到11.3%。作者证实,在生理环境下包覆DOX的聚合物胶束21 h内可将24.5%的DOX释放;而在p H= 5.0或10 mmol/L GSH(p H 7.4)的条件下,DOX的释放在相同时间内可分别达到62.8%与74.3%;在p H=5.0与10 mmol/L GSH的条件下,在10 h内可以将94.2%的DOX释放。MTT实验结果表明,该胶束对He La/RAW细胞的IC50值分别可达到0.75μg/m L与0.60μg/m L。
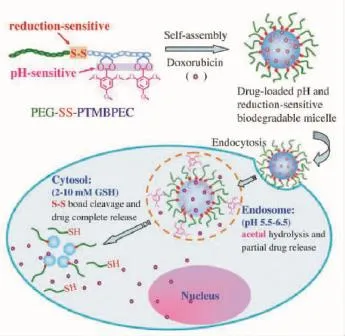
图22 基于GSH与pH双刺激响应的聚合物载药体系[75]Fig.22 GSH and p H dual responsive polymer nanocarriers and drug release[75]
2.4反应活性物种
相比于还原性刺激响应的解组装体系,对氧化剂刺激响应的聚合物组装体研究比较少,但是这一类组装体却非常适用于抗原递呈细胞(APCs)的抗原输运[76],其原因在于抗原递呈细胞(如树突细胞)核内体中高度的氧化环境(高H2O2浓度),由此Frechet等[77]将生物兼容性优良的葡聚糖用芳基硼酸酯加以修饰后得到相应的载体微球(如图23所示),构建了对H2O2刺激响应的解组装体系(Oxi-DEX)。作者证实在1 mmol/L的H2O2刺激下,这些微球对预装载药物释放的半衰期为36 min,而在没有H2O2的刺激时其半衰期大于1周。在随后的疫苗模型试验中,他们证实装载有卵清蛋白(OVA)的Oxi-DEX粒子对CD8+T细胞的表达是同样装载有OVA非氧化刺激响应性粒子的27倍。
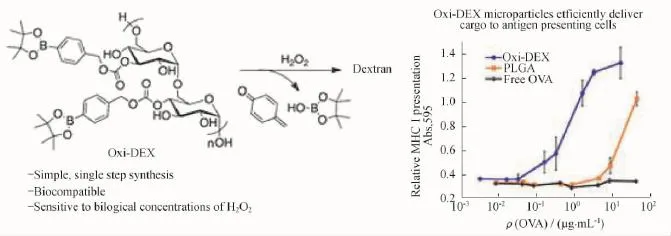
图23 基于H2O2刺激响应的聚合物载药体系[77]Fig.23 H2O2-responsive polymer nanocarriers and drug release[77]
许华平等[78]利用含有二硒键的嵌段共聚物,构建了氧化还原物种的刺激响应模式(如图24所示)。作者设计并合成了一种含有二硒键的新型聚氨酯(PUSeSe)为疏水链段,并在其两端接枝PEG为亲水链段,得到ABA型嵌段共聚物PEG-PUSeSe-PEG。后续的试验结果表明,PEG-PUSeSe-PEG在水相中可以自组装成非常稳定的胶束结构并包裹模型药物罗丹明B,同时该胶束对氧化还原刺激表现出优良的敏感度。在低浓度的过氧化氢(H2O2)或者谷胱甘肽(GSH)的作用下,聚合物链段中的二硒键断裂(生成硒酸或者硒醇),胶束结构崩塌,被胶束包裹的罗丹明B释放。这种多响应性的嵌段共聚物聚集体有望作为可控的药物输运体系,应用于化疗与放疗领域。
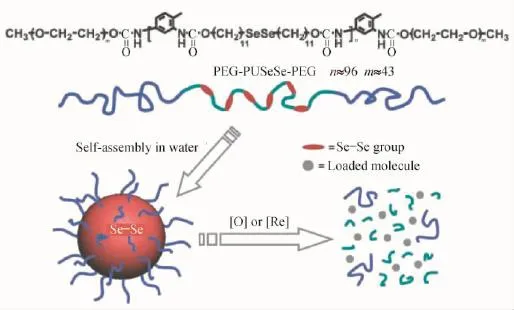
图24 对氧化还原物种刺激响应的二硒键嵌段共聚物体系[78]Fig.24 Redox dual-responsive assemblies formed from diselenide block copolymer[78]
随后,该课题组[79]在上述实验结果的基础上,成功开发了基于单线态氧(1O2)刺激响应的聚合物解组装体系(如图25所示)。作者在PEG-PUSeSe-PEG组装胶束中加入一种卟啉类光敏剂,在600~780 nm红光的照射下,体系中产生的单线态氧可以将二硒键氧化,从而导致胶束组装体解离,被包裹的Dox释放。
图25 对单线态氧(1O2)刺激响应的聚合物解组装体系[79]
Fig.25 Singlet oxygen(1O2)-responsive polymer assemblies[79]
3 结论与展望
针对传统刺激源响应的聚合物系统逐渐难以满足人们的特定胞内应用需求,因此,直接利用生物体细胞内已存在的相关物种作为刺激源,将会是刺激响应型聚合物自组装的一个重要发展方向,其优点在于原位刺激源的利用不会造成“附带”污染以及细胞损伤,同时还可以大大提高刺激响应的能力。现阶段人们已经相继开发出多种生物刺激源响应的聚合物自组装体系,包括生物大分子型的酶、DNA,小分子型的生物信号分子(NO,H2S),代谢物(ATP,CO2),生物活性分子(糖,生物硫醇)以及反应活性种(过氧化氢,单线态氧)等,这些研究大大推动了刺激响应聚合物自组装领域的发展,而对刺激响应性聚合物的应用使得聚合物科学与生物化学、分子生物学、细胞生物学等学科的联系更加紧密,从而为刺激响应性聚合物领域开拓出里程碑式的发展道路。需要指出的是,有很多生物相关物种虽然已经被证明对于生物体至关重要,但人们至今仍未开发出有效的策略对其进行检测与应用,比如一氧化碳(CO)、激素、各类维生素等。因此,人们在继续完善已有生物刺激源自组装体系的同时,也需要致力于对上述生物相关物种的刺激响应性大分子自组装体系的设计与开发。
[1] SCHATTLING P,JOCHUM F D,THEATO P,et al.Multi-stimuli responsive polymers:The all-in-one talents[J].Polym Chem,2014,5(1):25-36.
[2] WILLIAMS R J,SMITH A M,ULIJIN R V,et al.Enzyme-assisted self-assembly under thermodynamic control[J].Nat Nanotechnol,2009,4(1):19-24.
[3] ZHAIL.Stimuli-responsive polymer films[J].Chem Soc Rev,2013,42(17):7148-7160.
[4] SHIM M S,KWON Y J.Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications[J]. Adv Drug Delivery Rev,2012,64(11):1046-1058.
[5] PAEK K,YANG H,KIM B J.Efficient colorimetric p H sensor based on responsive polymer-quantum dot integrated graphene oxide[J].ACS Nano,2014,8(3):2848-2856.
[6] ROY D,CAMBRE J N,SUMERLIN B S.Future perspective and recent advances in stimuli-responsive materials[J].Prog Polym Sci,2010,35(1):278-301.
[7] STUART M A C,HUCK W T S,GENZER J,et al.Emerging applications of stimuli-responsive polymer materials[J]. Nat Mater,2010,9(2):101-113.
[8] TORCHILIN V P.Multifunctional,stimuli-sensitive nanoparticulate system for drug delivery[J].Nat Rev Drug Discov,2014,13(11):813-827.
[9] LIU F,URBAN M W.Recent advances and challenges in designing stimuli-responsive polymers[J].Prog Polym Sci,2010,35(1):3-23.
[10] LIU S Y,BILLINGHAM N C,ARMES S P.A schizophrenic water-soluble diblock copolymer[J].Angew Chem Int Ed,2001,40(12):2328-2331.
[11] RODRIGUEZ-HERNANDEZ J,LECOMMANDOUX S.Reversible inside-out micellization of p H-responsive and water-soluble vesicles based on polypeptide diblock copolymers[J].J Am Chem Soc,2005,127(7):2026-2027.
[12] LI G Y,SHI L Q,HUANG N.Formation of complex micelles with double-responsive channels from self-assembly of two diblock copolymer[J].Angew Chem Int Ed,2006,45(30):4959-4962.
[13] DU J Z,DU X J,WANG J,et al.Tailormade dual p H-sensitive polymerdoxorubicin nanoparticless for efficient anticancer drug delivery[J].J Am Chem Soc,2011,133(44):17560-17563.
[14] NAPOLO A,VALENTINI M,HUBBEL J A,et al.Oxidation-responsive polymer vesicles[J].Nat Mater,2004,3(3):183-189.
[15] MA N,XU Y,ZHANG X,et al.Dual redox responsive assemblies formed from diselenide block copolymers[J].J Am Chem Soc,2010,132(2):442-443.
[16] LI Y,WANG Y G,GAO J M,et al.Chaotropic-anion-induced supramolecular self-assembly of ionic polymeric micelles[J]. Angew Chem Int Ed,2014,53(31):8212-8216.
[17] LI Y T,LOKITZ B S,McCORMICK C L.Thermally responsive vesicles and their structural locking through polyelectrolyte complex formation[J].Angew Chem Int Ed,2006,45(35):5792-5795.
[18] LUTZ J F,AKDEMIR O,HOTH A.Point by point comparison of two thermoresponsive polymers exhibiting a similar LCST:Is the age of poly(NIPAM)over?[J].J Am Chem Soc,2006,128(40):13046-13047.
[19] YAN Q,YUAN J Y,YIN Y W,et al.Voltage-responsive vesicles based on orthogonal assembly of two homopolymers[J]. J Am Chem Soc,2010,132(27):9268-9270.
[20] JIANG J Q,TONG X,ZHAO Y.A new design for light-breakable polymer micelles[J].J Am Chem Soc,2005,127(23):8290-8291.
[21] FOMINA N,MCFEATIN,ALMUTAIRI A,et al.UV and near-IR triggered release from polymeric nanoparticles[J].J Am Chem Soc,2010,132(28):9540-9542.
[22] YAN B,BOYER J C,ZHAO Y,et al.Near-infrared light-triggered dissociation of block copolymer micelles using upconverting nanoparticles[J].J Am Chem Soc,2011,133(49):19714-19717.
[23] TAN X Y,LI B B,ZHANG K,et al.Light-triggered,self-immolative nucleic acid-drug nanostructures[J].J Am Chem Soc,2015,137(19):6112-6115.
[24] WANG X R,HU J M,LIU S Y,et al.Reversibly switching bilayer permeability and release modules of photochromic polymersomes stabilized by cooperative noncovalent interactions[J].J Am Chem Soc,2015,137(48):15262-15275.
[25] LIU G N,ZHANG G F,LIU S Y,et al.Hyperbranched self-immolative polymers(hSIPs)for programmed payload delivery and ultrasensitive detection[J].J Am Chem Soc,2015,137(36):16645-16655.
[26] YAN Q,ZHAO Y.Block copolymer self-assembly controlled by the“green”gas stimulus of carbon dioxide[J].Chem Commun,2014,50(79):11631-11641.
[27] de BOER B,STALMACH U,HADZIIOANNOU G,et al.Supramolecular self-assembly and opto-electronic properties of semiconducting block copolymers[J].Polymer,2001,42(21):9097-9109.
[28] WOLKOFF P,SCHNEIDER T,SCHUNK H,et al.Risk in cleaning:Chemical and physical exposure[J].Sci Total Environ,1998,215(1-2):135-136.
[29] MURA S,NICOLAS J,COUVREUR P.Stimuli-responsive nanocarriers for drug delivery[J].Nat Mater,2013,12(11):991-1003.
[30] AMIR R J,ZHONG S,HAWKER C J,et al.Enzymatically triggered self-assembly of block copolymers[J].J Am Chem Soc,2009,131(39):13949-13951.
[31] MEERS P.Enzyme-activated targeting of liposomes[J].Adv Drug Delivery Rev,2001,53(31):265-272.
[32] MOLLA M R,PRIYAA P,THAYUMANAVAN S.Protein-induced supramolecular disassembly of amphiphilic polypeptide nanoassemblies[J].J Am Chem Soc,2015,137(23):7286-7289.
[33] GUO J,ZHUANG J M,THAYUMANAVAN S,et al.Protein and enzyme gated supramolecular disassembly[J].J Am Chem Soc,2014,136(6):2220-2223.
[34] AZAGARSAMY M A,YESILYURT V,THAYUMANAVAN S.Disassembly of dendritic micellar containers due to protein binding[J].J Am Chem Soc,2010,132(13):4550-4551.
[35] RAO J Y,KHAN A.Enzyme sensitive synthetic polymer micelles based on the azobenzene motif[J].J Am Chem Soc,2013,135(38):14056-14059.
[36] WANG C,CHEN Q S,ZHANG X,et al.An enzyme-responsive polymeric superamphiphile[J].Angew Chem Int Ed,2010,49(46):8612-8615.
[37] SEEMAN N C.DNA in a material world[J].Nature,2003,421(6921):427-431.
[38] ALDAYE F A,PALMER A L,SLEIMAN H F.Assembling Materials with DNA as the guide[J].Science,2008,321 (5897):1795-1799.
[39] MCLAUGHLIN C K,HAMBLIN G D,SLEIMAN H F,et al.Supramolecular DNA assembly[J].Chem Soc Rev,2011,40(12):5647-5656.
[40] EDWARDSON T G W,CARNEIRO K M M,SLEIMAN H F,et al.Site-specific positioning of dendritic alkyl chains onDNA cages enables their geometry-dependent self-assembly[J].Nature Chemistry,2013,5(10):868-875.
[41] BUJOLD K E,FAKHOUTY J,SLEIMAN H F,et al.Sequence-responsive unzipping DNA cubes with tunable cellular uptake profiles[J].Chem Sci,2014,5(6):2449-2455.
[42] NIU L N,CHEN Y Z,YANG Q Z,et al.Design strategies of fluorescent probes forselective detection among biothiols[J]. Chemical Society Reviews,2015,44(17):6143-6160.
[43] LI L,ROSE P,MOORE,P K.Hydrogen sulfide and cell signaling[J].Annu Rev Pharmacol Toxicol,2011,51(51):169-187.
[44] LI L,MOORE,P K.Putative biological roles of hydrogen sulfide in health and disease:A breath of not so fresh air?[J]. Trends Pharmacol Sci,2008,29(2):84-90.
[45] YAN Q,SANG W.H2S gasotransmitter-responsive polymer vesicles[J].Chem Sci,2016,7(3):2100-2105.
[46] RICCIO D A,SCHOENFISCH M H.Nitric oxide release:Part I.Macromolecular scaffolds[J].Chem Soc Rev,2012,41 (10):3731-3741.
[47] CONESKI P N,SCHOENFISCH M H.Nitric oxide release:PartⅢ.Measurement and reporting[J].Chem Soc Rev,2012,41(10):3753-3758.
[48] MOCELLIN S,BRONTE V,NITTI D.Nitric oxide,a double edged sword in cancer biology:Searching for therapeutic opportunities[J].Med Res Rev,2007,27(3):317-352.
[49] GLADWIN M T,KIM-SHAPIRO D B.Vascular biology:Nitric oxide caught in traffic[J].Nature,2012,491(7424):344-345.
[50] HU J M,WHITTAKER M R,Davis T P,et al.Biomimetic polymers responsive to a biological signaling molecule:Nitric oxide triggered reversible self-assembly of single macromolecular chains into nanoparticles[J].Angew Chem Int Ed,2014,53(30):2583-2589.
[51] GUO Z Q,SONG NR,YOON J Y,et al.A benzobisimidazolium-based fluorescent and colorimetric chemosensor for CO2[J].J Am Chem Soc,2012,134(43):17846-17849.
[52] WANG H,CHEN D D,DONG Y P.A fluorescent probe with an aggregation-enhanced emission feature for real-time monitoring of low carbon dioxide levels[J].J Mater Chem C,2015(3):7621-7626.
[53] DANSBY-SPARKS R N,JIN J,XUE Z L,et al.Fluorescent-dye-doped sol-gel sensor for highly sensitive carbon dioxide gas detection below atmospheric concentrations[J].Anal Chem,2010,82(2):593-600.
[54] GUTKNECHT J,BISSON M A,TOSTESON F C.Diffusion of carbon dioxide through lipid bilayer membranes:Effects of carbonic anhydrase,bicarbonate,and unstirred layers[J].J Gen Physiol,1977,69(6):779-784.
[55] TOUR J M,KITTRELL C,COLVIN V L.Green carbon as a bridge to renewable energy[J].Nat Mater,2010,9(11):871-874.
[56] YAN Q,ZHOU R,YUAN J Y,et al.CO2-responsive polymeric vesicles that breathe[J].Angew Chem Int Ed,2011,50 (21):4923-4927.
[57] YAN Q,WANG J B,YUAN J Y,et al.Breathing polymersomes:CO2-tuning membrane permeability for size-selective release,separation,and reaction[J].Angew Chem Int Ed,2013,52(19):5070-5073.
[58] YAN Q,ZHAO Y.Polymeric microtubules that breathe:CO2-driven polymer controlled-self-assembly and shape transformation[J].Angew Chem Int Ed,2013,52(38):9948-9951.
[59] YAN Q,ZHAO Y.CO2-stimulated diversiform deformations of polymer assemblies[J].J Am Chem Soc,2013,135(44):16300-16303.
[60] JIE K C,ZHOU Y J,HUANG F H,et al.CO2-responsive pillar[5]arene-based molecular recognition in water:Establishment and application in gas-controlled self-assembly and release[J].J Am Chem Soc,2015,137(33):10472-10475.
[61] BISWAS S,KINBABA K,AIDA T,et al.Biomolecular robotics for chemomechanically driven guest delivery fuelled by intracellular ATP[J].Nat Chem,2013,5(7):613-620.
[62] OKURO K,SASAKI M,AIDA T.Boronic acid-appended molecular glues for ATP-responsive activity modulation of enzymes[J].J Am Chem Soc,2016,DOI:10.1021/jacs.6b02664.
[63] MO R,JIANG T Y,GU Z,et al.ATP-triggered anticancer drug delivery[J].Nature Communications,2014,5(1):3364.
[64] YAN Q,ZHAO Y.ATP-triggered biomimetic deformations of bioinspired receptor-containing polymer assemblies[J]. Chem Sci,2015,6(7):4343-4349.
[65] YU G C,ZHOU J,HUANG F H,et al.Cationic pillar[6]arene/ATP host-guest recognition:Selectivity,inhibition of ATP hydrolysis,and application in multidrug resistance treatment[J].Chem Sci,2016,DOI:10.1039/C6SC00531D.
[66] XU X D,LIN B B,ZHUO R X,et al.Biological glucose metabolism regulated peptide self-assembly as a simple visual biosensor for glucose detection[J].Macromol Rapid Commun,2012,33(5):426-431.
[67] RYU J H,JIWPANICH S,THAYUMANAVAN S,et al.Surface-functionalizable polymer nanogels with facile hydrophobic guest encapsulation capabilities[J].J Am Chem Soc,2010,132(24):8246-8247.
[68] RYU J H,CHACKO R T,THAYUMANAVAN S,et al.Self-cross-linked polymer nanogels:A versatile nanoscopic drug delivery platform[J].J Am Chem Soc,2010,132(48):17227-17235.
[69] JIWPANICH S,RYU J H,THAYUMANAVAN S,et al.Noncovalent encapsulation stabilities in supramolecular nanoassemblies[J].J Am Chem Soc,2010,132(31):10683-10685.
[70] ZHAO W,ZHANG H,SHI J,et al.A glucose-responsive controlled release of insulin system based on enzyme multilayers-coated mesoporous silica particles[J].Chem Commun,2011,47(33):9459-9461.
[71] GUO Q Q,ZHANG T Q,LI C X,et al.Block versus random amphiphilic glycopolymer nanopaticles as glucose-responsive vehicles[J].Biomacromolecules,2015,16(10):3345-3356.
[72] KIM H,KANG Y J,KIN T,et al.Monosaccharide-responsive release of insulin from polymersomes of polyboroxole block copolymers at neutral p H[J].J Am Chem Soc,2012,134(9):4030-4033.
[73] CHENG R,FENG F,ZHONG Z,et al.Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery[J].J Controlled Release,2011,152(1):2-12.
[74] KUPPUSAMY P,LI H,MITCHELL J B,et al.Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels[J].Cancer Res,2002,62(1):307-312.
[75] CHEN W,ZHONG P,ZHONG Z Y,et al.Redox and p H-responsive degradable micelles for dually activatedintracellular anticancer drug release[J].Journal of Controlled Release,2013,169(3):171-179.
[76] WANG H,WANG X,MANNERS I,et al.Redox-mediated synthesis and rncapsulation of inorganic nanoparticles in shell-cross-linked cylindrical polyferrocenylsilane block copolymer micelles[J].J Am Chem Soc,2008,130(39):12921-12930.
[77] BROADERS K E,GRANGHE S,FRECHET J M.A Biocompatible oxidation-triggered carrier polymer with potential in therapeutics[J].J Am Chem Soc,2011,133(4):756-758.
[78] MA N,XU H P,ZHANG X,et al.Dual redox responsive assemblies formed from diselenide block copolymers[J].J Am Chem Soc,2010,132(2):442-443.
[79] HAN P,LI S C,XU H P,et al.Red light responsive diselenide-containing block copolymer micelles[J].J Mater Chem B,2013,1(6):740-743.
Biological Stimuli-Responsive Polymers and Their Controllable Self-Assembly
ZHANG Jian, MA Ming-xuan, YAN Qiang
(State Key Laboratory of Molecular Engineering of Polymers,Department of Macromolecular Science,Fudan University,Shanghai 200433,China)
Biological stimuli-responsive polymer self-assembly systems are crucial to the development of smart macromolecules.This review summarizes the research status on the polymer assemblies that can respond to the biomacromolecular-type and small-molecular-type stimulants.Furthermore,according to the difference of stimulation categories,the great progress of this filed is introduced and the valuable perspective of bioresponsive polymers is propsed.
biological stimulus;responsive polymers;controllable self-assembly
O63
A
1008-9357(2016)02-0115-019DOI: 10.14133/j.cnki.1008-9357.2016.02.001
2016-05-12
中组部青年千人计划项目(KHH17171002)
张 建(1987-),男,山西晋中人,博士,研究方向为智能聚合物的设计与功能应用。E-mail:zhangjian507@fudan.edu.cn
闫 强(1985-),男,博士,教授,博导,2014年入选中组部第6批青年千人计划,研究方向为刺激响应性聚合物的设计与功能开发、智能大分子纳米器件与仿生细胞。E-mail:yanq@fudan.edu.cn
