氟喹酮的合成工艺改进
2016-10-25徐盼云陈志卫
徐盼云, 陈志卫
(浙江工业大学 药学院 绿色制药技术与装备教育部重点实验室,浙江 杭州 310014)
·制药技术·
氟喹酮的合成工艺改进
徐盼云, 陈志卫*
(浙江工业大学 药学院 绿色制药技术与装备教育部重点实验室,浙江 杭州310014)
以靛红酸酐为起始原料,经硝化、还原上保护、氨解开环、环合、卤素交换和脱保护共七步反应合成了氟喹酮(7),其结构经1H NMR,13C NMR和ESI-MS确证。研究了硝化试剂、溶剂、催化剂等对7收率的影响,结果表明:在使用浓硝酸作为硝化试剂,甲苯,乙腈等为溶剂,Raney-Ni, TBAB等为催化剂时,总收率可达58%,纯度达99%以上。
肌松药; 氟喹酮; 靛红酸酐; 药物合成
氟喹酮(7) 是一种对中枢神经起效的肌肉松弛药,依据上世纪70 年代催眠药安眠酮进行结构修饰得到的[1-2]。7由日本的田边株式会社所开发,于1983年在日本首次上市。是临床主要作用于脊上位中枢较广泛的部位而使肌肉紧张性亢进状态缓解[3]。与已知的肌松药相比,7的副作用和毒性都较小,具有很好的安全性[4]。
目前7的合成方法主要有四种:(1)以N-(2-氨基-5-硝基苯甲酰基)邻甲苯胺为原料,与氟代乙酰氯缩合后在醋酐作用下环合生成2-氟甲基-3-邻甲苯基-6-硝基-4(3H)-喹唑啉酮,最后用Pd-C氢化还原生成7[5](总收率66.6%)。该路线主要缺点是使用的氟代乙酰氯毒性大且价格昂贵;(2)以N-(2-氨基-5-硝基苯甲酰基)邻甲苯胺为原料,与氯乙酰氯直接环合生成2-氯甲基-3-邻甲苯基-6-硝基-4(3H)-喹唑啉酮,再进行氟交换和还原反应生成7[6](总收率41.1%);(3)以N-(2-氨基-5-硝基苯甲酰基)邻甲苯胺为原料,与氯乙酰氯环合,再进行硝基还原后上保护,接着进行氟交换和水解脱保护得到7[7](总收率28.0%);(4)先将N-(2-氨基-5-硝基苯甲酰基)邻甲苯胺进行硝基还原后上保护,再与氯乙酰氯环合,接着进行氟交换和水解脱保护生成7[8](总收率33.6%)。后三种方法主要缺点是总收率较低。

Scheme 1
本文以靛红酸酐(1)为起始原料,经硝化[9-12]、还原上保护、氨解开环、环合、卤素交换和脱保护等七步反应合成7(Scheme 1),总收率提高至60.0%。该工艺具有以下优点:(1)起始原料价廉易得,降低了生产成本;(2) 避免使用原料氟代乙酰氯;(3) 氟交换反应中使用四丁基溴化铵作为相转移催化剂,提高了该步的转化率。
1 实验部分
1.1仪器与试剂
WRS-1A型数字熔点仪(温度未校正);Varian-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Trace DSQ FINNIGSH型质谱仪。
1,上海有朋化工有限公司;其余所用试剂均为分析纯或化学纯。
1.2合成
(1) 5-硝基靛红酸酐(2)的合成
在三口瓶中加入浓硫酸100 mL,搅拌下于0~10 ℃缓慢加入1 30.0 g(0.18 mol),搅拌使其溶解后滴加65%硝酸21.4 g(0.22 mol),滴毕(1 h),反应1 h。将反应液缓慢倒至300 mL冰水中,搅拌20 min(固体充分析出),过滤,滤饼用水洗涤,干燥得黄色固体2 44.8 g,纯度98.4%(HPLC),收率98.0%, m.p. 248.8~249.5 ℃(224~232 ℃[9]);1H NMRδ: 7.24(d,J=8.0 Hz, 1H), 8.43~8.50(m, 1H), 8.49~8.50(d,J=8.0 Hz, 1H), 12.24(s, 1H); MS(ESI)m/z: 209 {[M+H]+}。
(2) 5-乙酰氨基靛红酸酐(3)的合成
在500 mL高压釜内依次加入2 20.0 g(96.0 mmol),四氢呋喃 200 mL和雷尼镍 1.0 g,充氮气置换空气三次,检查气密性后再用氢气置换氮气三次,通氢气维持压力为1.0 Mpa,于40 ℃反应4 h。过滤,向滤液中加入醋酐9.8 g(96.0 mmol),反应1 h。减压浓缩,残余物用乙醇(50 mL)重结晶得白色固体3 20.0 g,纯度98.0%(HPLC),收率95.0%, m.p. 220.6~221.9 ℃;1H NMRδ: 2.23(s, 3H), 7.08(d,J=8.8 Hz, 1H), 7.32~7.38(m, 1H), 7.77(d,J=8.8 Hz, 1H), 8.64(s, 1H), 12.14(s, 1H); MS(ESI)m/z: 221{[M+H]+}。
(3)N-(2-氨基-5-乙酰氨基苯甲酰基)邻甲苯胺(4)的合成
在三口瓶中依次加入3 8.0 g(36.3 mmol),邻甲苯胺3.9 g(36.3 mmol)和乙醇 50 mL,搅拌下回流(85 ℃)反应4 h。冷却至室温,减压浓缩,残余物用20 mL异丙醇重结晶得白色固体4 9.8 g,纯度98.8%(HPLC),收率95%, m.p. 201.6~211.9 ℃(215~217 ℃[8]);1H NMRδ: 1.99(s, 3H), 2.23(s, 3H), 5.98(s, 2H), 6.68(d,J= 8.8 Hz, 1H), 7.10~7.24(m, 3H), 7.32~7.38(m, 2H), 7.77(d,J=2.0 Hz, 1H), 9.64(s, 2H); MS(ESI)m/z: 284 {[M+H]+}。
(4) 6-乙酰氨基-2-氯甲基-3-(2-甲基苯基)-4(3H)-1,3-二氮杂萘酮(5)的合成
在三口瓶中依次加入冰醋酸 50 mL和4 8.0 g(28.2 mmol),搅拌下缓慢滴加氯乙酰氯3.3 g(28.2 mmol),滴毕(20 min),回流反应4 h。冷却至室温,减压浓缩得灰白色固体,用乙醇(20 mL)重结晶得淡黄色固体5 9.6 g,纯度97.5%(HPLC),收率92.0%, m.p. 208.5~210.0 ℃(219~221 ℃[8]);1H NMRδ: 1.68(s, 3H), 2.13(s, 3H), 4.11(d,J=12.0 Hz, 1H), 4.28(d,J= 11.6 Hz, 1H), 7.26~7.45(m, 4H), 7.76(d,J= 9.2 Hz, 1H), 8.09(d,J=2.4 Hz, 1H), 8.60(d,J= 9.2 Hz, 1H), 8.72(s, 1H); MS(ESI)m/z: 342 {[M+H]+}。
(5) 6-乙酰氨基-2-氟甲基-3-(2-甲基苯基)-4(3H)-1,3-二氮杂萘酮 (6)的合成
在圆底烧瓶中依次加入5 7.5 g(22 mmol),无水乙腈 40 mL、活化氟化钾 3.83 g(66 mmol)和10 mol%四丁基溴化铵 0.71 g(2.2 mmol), 搅拌下回流反应4 h。冷却至室温,加入二氯甲烷30 mL,过滤(回收过量的KF),滤液减压浓缩得黄色固体粗品6.63 g(粗收率85.6%),用乙醇(20 mL)重结晶得黄色固体6 6.3 g,纯度95.6%(HPLC),收率90.2%, m.p. 248.3~250.3 ℃(239~242 ℃[8]);1H NMR(400 MHz, CDCl3)δ: 1.61(s, 3H), 2.11(s, 3H), 4.83~5.01(m, 2H), 7.20~7.42(m, 4H), 7.80(d,J=8.0 Hz, 1H), 8.11(s, 1H), 8.61~8.76(m, 2H); MS(ESI)m/z: 326 {[M+H]+}。
(6) 7的合成
在烧瓶中依次加入氢氧化钠 1.23 g(30.8 mol), 15 mL水和30 mL乙醇,搅拌使其溶解;加入6 5.0 g(15.4 mmol),于70 ℃反应2 h。减压浓缩回收乙醇,用二氯甲烷萃取,合并有机层,减压浓缩得棕色黏稠油状液体,用异丙醇(7 mL)重结晶得黄色固体7 3.7 g,纯度99.0%(HPLC),收率85.0%, m.p. 193.2~195.7 ℃(194~196 ℃[8]);1H NMRδ: 2.01(s, 3H), 4.81~5.02(m, 2H), 5.85(s, 2H), 7.15(dd,J=2.8 Hz, 8.8 Hz, 1H), 7.23(d,J=2.8 Hz, 1H), 7.35~7.44(m, 4H), 7.51(d,J=8.8 Hz, 1H); MS(ESI)m/z: 284 {[M+H]+}。
2 结果与讨论
2.12的合成条件优化
在2的合成中,考察了浓硝酸和发烟硝酸对反应的影响,结果见表1。由表1可见,与发烟硝酸相比,浓硝酸为硝化试剂时,产率略有降低,但出于成本和安全考虑,选择硝化剂为浓硝酸。
2.23的合成条件优化
考察了催化剂及其用量对3收率的影响,结果见表2。由表2可见,同等用量时Raney-Ni的催化效果不如钯碳,Raney-Ni的用量加至5%,催化效果优于钯碳,且此时成本仍低于钯碳,选择Raney-Ni作为催化氢化催化剂。

表1 硝化试剂对反应的影响*
*温度0~10 ℃,硝化试剂的滴加时间为1 h,r=n(1) ∶n(硝化试剂)=1.0 ∶1.2。

表2 还原反应的考察结果*
*乙酸酐1 eq.,温度0~5 ℃,乙酸酐的滴加时间为1 h,还原温度40 ℃,压力1×106Pa,时间为24 h。
2.34的合成条件优化
在4的合成中,分别考察了溶剂和邻甲苯胺的用量对反应的影响。
(1) 溶剂
考察了甲苯和乙醇对反应的影响(未用乙醇洗,未进行重结晶),结果见表3。由表3可见,甲苯作溶剂,反应温度高,原料反应较完全,收率较高,选择甲苯为溶剂。
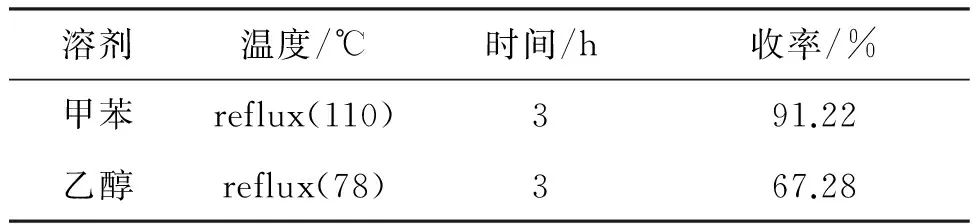
表3 溶剂对反应的影响*
*y=n(3) ∶n(邻甲苯胺)=1.0 ∶2.0。
(2) 邻甲苯胺用量
考察了邻甲苯胺的用量对反应的影响,反应结束进行抽滤,用乙醇洗去多硝基取代副产物,未进行重结晶,结果见表4。由表4可见, 邻甲苯胺的用量为1.5 eq.时的粗收率最高(96.0%)。
2.46的合成条件优化
(1) 催化剂种类
KF是无机物,不溶于所选有机溶剂,为了使KF和5充分接触,考察不同的相转移催化剂。结果见表5。由表5可见,TBAB催化的粗产率远远大于18-冠-6,且18-冠-6毒性较大,刺激性较强,对环境不友好。综合考虑选择TBAB作为氟交换反应的催化剂。

表4 邻甲苯胺用量对反应的影响*
*甲苯为溶剂。

表5 催化剂对反应的影响*
*DMF为溶剂,反应温度130 ℃
(2) 溶剂
选择极性较强的溶剂,以5%TBAB为催化剂,结果见表6。由表6可见,以乙腈作溶剂的收率最高,所以选择乙腈作为氟交换的溶剂。
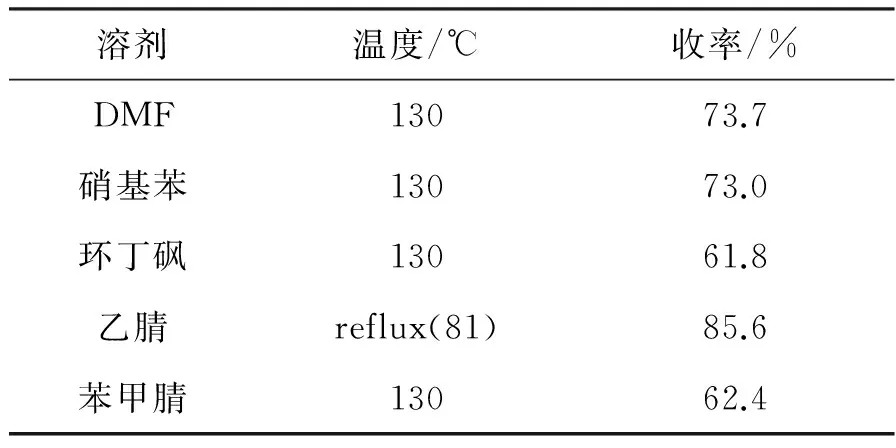
表6 溶剂对反应的影响
以廉价的靛红酸酐为起始原料,经7步反应合成氟喹酮(7)。研究了硝化试剂、溶剂、催化剂等对7收率的影响,结果表明:在使用浓硝酸作为硝化试剂,甲苯,乙腈等作为溶剂,Raney-Ni,TBAB等作为催化剂时,总收率可达58%,纯度达99%。该工艺条件具有反应条件温和、操作简便,总收率较高等优点,有利于工业化生产。
[1]Junichi T, Yoshihisa Y, Toyonari O,etal. Studies on biologically active halogenated compounds.1.synthesis and central nervous system depressant activity of 2-(fluoromethyl)-3-aryl-4(3H)-quinazolinone derivatives[J].J Med Chem,1979,22(1):95-99.
[2]Ochiai T, Ishida R. Pharmacological studies on 6-amino-2-fluoromethyl-3-(O-tolyl)-4(3H)-quinazolinone(afloquanone),a new centrally acting muscle relaxant.(II) Effects on the spinal reflex potential and the rigidity.[J].Japan J Pharmacol,1981,31:491-501.
[3]Ochiai T, Yamamura M, Kudo Y. General pharmacology of 6-amino-2-fluoro-methyl-3-(o-tolyl)-4(3H)-quinazolinone(afloqualone),a new centrally acting muscle relaxant.I.Effects on the central nervous system[J].Nippon Yakurigaku Zasshi,1981,78(4):347-357.
[4]Kojima M, Kudo Y, Ishida R. Effects of afloqualone,a centrally acting muscle relaxant,on the sleep-wakefulness cycle in cats with chronically implanted electrodes(author′s transl)[J].Nihon yakurigaku zasshi.Folia pharmacologica Japonica,1981,78(5):471-482.
[5]Ichizo I, Toyonari O, Yoshihisa Y.etal. 2-fluoromethyl-3-tolyl-6-amino- 4(3H)-quinazolinone:US 3 966 731[P].1976.
[6]井上一三. 喹唑啉酮衍生物的制法:JP 51 105 083[P].1976.
[7]井上一三. 喹唑啉酮衍生物的制法:JP 51 105 082[P].1976.
[8]陈芬儿. 有机药物合成法[M].上海:中国医药科技出版社,1999,440-444.
[9]Das J, Kumar M S, Subrahmayam D,etal. Substituent activity relationship studies on new azolo benzoxazepinyl oxazolidinones[J].Bioorg Med Chem,2006,14(23)8032-8042.
[10]Kharul R K, Prajapati P N, Thorave A,etal. Effective synthesis of 1,5-disubstituted-2,1-benzisothiazol-3(1H)-ones[J].Syn Commun,2011,41(22):3265-3279.
[11]Shakhidoyatov K M, Arslanova O N, Eshimbetov Z,etal. Synthesis of 3,1-benzoxazine-2,4-diones and their reaction with electrophilic reagents[J].Dokl Akad Nauk,1989,7:41-42.
[12]Adepu R, Rajnikanth S, Meda C T,etal. Facile assembly of two 6-membered fusedN-heterocyclic rings:A rapid access to novel small moleculesviaCu-mediated reaction[J].Chem Commun,2013,49(2):190-192.
Process Improvement on Synthesis of Afloqualone
XU Pan-yun,CHEN Zhi-wei*
(Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education, College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China)
Afloqualone was synthesized from isatoic anhydrideviaseven-step reaction including nitration, reduction, protection, solasodamine, annulation, halogen exchange and deprotection. The structure was confirmed by1H NMR,13C NMR and ESI-MS. The influence of nitration reagents, solvents, catalysts on the yield of 7 were studied. The results showed that when using nitric acid as nitrating agent, toluene, acetonitrile as the solvent, Raney-Ni, TBAB as the catalyst, the total yield can reach 58%, the purity can reach 99%.
afloquanone; muscle relaxant activity; isatoic anhydride; drug synthesis
2015-11-23;
2016-06-23
徐盼云(1991-),男,汉族,浙江东阳人,硕士,主要从事医药中间体的合成研究。
陈志卫,副教授, Tel. 0571-88871087, E-mail: chenzhiwei@zjut.edu.cn
O621.3; R914.5
ADOI: 10.15952/j.cnki.cjsc.1005-1511.2016.09.15386
