氮杂环丁烷衍生物的合成及其抗肿瘤活性
2016-10-25赵淑艳左爱学毛泽伟
赵淑艳, 左爱学, 毛泽伟
(1. 承德护理职业学院,河北 承德 067000; 2. 云南中医学院 中药学院,云南 昆明 650500)
氮杂环丁烷衍生物的合成及其抗肿瘤活性
赵淑艳1, 左爱学2, 毛泽伟2
(1. 承德护理职业学院,河北 承德067000; 2. 云南中医学院 中药学院,云南 昆明650500)
以3-羟基氮杂环丁烷盐酸盐为原料,经取代、官能团转化制得3-氨基氮杂环丁烷化合物(3); 3经衍生化合成了10个3-氨基氮杂环丁烷衍生物(4a~4j),其中4f~4j为新化合物,其结构经1H NMR,13C NMR和HR-MS(ESI-TOF)表征。采用MTT法初步测试了化合物的体外抗肿瘤活性。结果表明:4h 对A549表现出较强的细胞毒活性(IC50=8.06 μmol·L-1)。
3-羟基氮杂环丁烷; 氮杂环丁烷; 衍生物; 合成; 抗肿瘤活性
3-氨基氮杂环丁烷类化合物具有良好的抗菌、消炎和细胞毒等活性,广泛用于药物研发及药物合成,如1-二苯甲基-3-氨基氮杂环丁烷和1-二苯甲基-3-羟基-3-氨甲基氮杂环丁烷用于合成抗菌、抗抑郁类药物[1-3]。鉴于3-氨基氮杂环丁烷良好的生理活性,近年来对该类化合物进行了大量的构效关系研究[4-8]。苯乙酮类化合物具有广泛的生物活性,是非常重要的医药中间体和原料。在前期研究中,我们将苯乙酮和氮杂环丁烷片段连接起来,通过官能团转化对氮杂环丁烷衍生物的合成方法进行了研究[9],在此基础上,本文希望制得具有更优药理活性的3-氨基氮杂环丁烷类酰胺及磺酰胺衍生物。
以4-氟苯乙酮与3-羟基氮杂环丁烷盐酸盐为原料,经取代、官能团转化反应制得3-氨基氮杂环丁烷化合物(3);再对氨基进行衍生化,合成了10个氮杂环丁烷衍生物 (4a~4j)(Scheme 1),其中4f~4j为新化合物,其结构经1H NMR,13C NMR和HR-MS(ESI-TOF)表征。初步测试了4 的体外细胞毒活性。结果表明:4h 对肿瘤细胞株A549表现出较强的细胞毒活性(IC50=8.06 μmol·L-1),可做进一步研究。
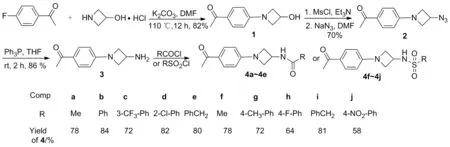
Scheme 1
1 实验部分
1.1仪器与试剂
Bruker AM 300型核磁共振仪(CDCl3为溶剂,TMS为内标);AutoSpec Premier P776型双聚焦三扇型磁质谱仪;二氧化碳培养箱;Biotek型Epoch连续波长酶标仪;RPMI-1640培养基。
所用试剂均为分析纯。
1.2合成
(1) 4-(3-羟基氮杂环丁基)苯乙酮(1)的合成
将4-氟苯乙酮1.38 g(10 mmol), 3-羟基氮杂环丁烷盐酸盐1.64 g(15 mmol)和无水DMF 30 mL加入50 mL圆底烧瓶中,搅拌下加入K2CO34.14 g(30 mmol),加毕,于110 ℃反应12 h(TLC检测)。倒入冷水中(100 mL),搅拌10 min。抽滤,滤饼用20%乙醇水溶液洗涤,干燥得淡灰色固体 1,收率82%, m.p.140~142 ℃;1H NMRδ: 7.83(d,J=7.2 Hz, 2H), 6.37(d,J=6.0 Hz, 2H), 4.80(s, 1H), 4.27~4.22(m, 2H), 3.84~3.79(dd,J=4.2 Hz, 4.5 Hz, 2H), 2.80(s, 1H), 2.49(s, 3H);13C NMRδ:196.72, 154.03, 130.43, 126.48, 110.15, 62.43, 61.08, 26.00。
(2) 4-(3-叠氮基氮杂环丁基)苯乙酮(2)的合成
将1 1.91 g(10 mmol),无水DCM 30 mL和三乙胺2 ml加入圆底烧瓶中,冰水浴冷却下缓慢滴加MsCl 1 mL,滴毕,升至室温反应12 h;缓慢滴加饱和NaHCO3溶液10 mL,滴毕,用DCM萃取,有机相用水洗涤后经无水硫酸钠干燥,真空浓缩,残余物用无水DMF溶解(20 mL),加入叠氮化钠2.00 g,于90 ℃反应24 h。冷却至室温,加乙酸乙酯40 mL,用水洗涤,有机相经无水硫酸钠干燥,真空浓缩,残余物经硅胶柱层析(洗脱剂:DCM)纯化得黄褐色黏稠液体,两步收率70%;1H NMRδ: 7.84(d,J=8.7 Hz, 2H), 6.38(d,J=8.4 Hz, 2H), 4.43~4.40(m, 1H), 4.25(t,J=8.1 Hz, 2H), 3.91~3.86(dd,J=4.8 Hz, 4.8 Hz, 2H), 2.48(s, 3H);13C NMRδ: 196.45, 153.40, 130.50, 130.36, 127.24, 110.31, 57.69, 50.19, 26.08。
(3) 3的合成
将2 1.08 g(5 mmol)加入圆底烧瓶中,用THF 20 mL溶解,搅拌下于室温分三批加入三苯基膦2.62 g(10 mmol),加毕,反应5 h(TLC检测)。真空浓缩反应液,残余物经硅胶柱层析(洗脱剂:A=DCM/CH3OH=50/1,V/V)纯化得淡黄色固体3,收率86%, m.p.142~144 ℃;1H NMRδ: 7.64(d,J=7.2 Hz, 2H), 6.37(d,J=6.9 Hz, 2H), 4.25(t,J=7.5 Hz, 2H), 4.01~3.96(m, 1H), 3.61~3.57(dd,J=5.1 Hz, 5.1 Hz, 2H), 2.64(s, 2H), 2.49(s, 3H);13C NMRδ: 196.43, 154.10, 131.98, 130.37, 128.60, 110.04, 61.50, 43.63, 26.02。
(4) 4a~4j的合成通法
将3 100 mg,无水DCM 10 mL和三乙胺0.5 mL加入圆底烧瓶中,冰水浴冷却下缓慢滴加酰氯或磺酰氯0.2 mL,滴毕,升至室温反应12 h。缓慢滴加饱和NaHCO3溶液至无气泡产生,用水(30 mL)洗涤,用二氯甲烷萃取,有机相用无水硫酸钠干燥,真空浓缩,残余物经硅胶柱层析(洗脱剂:DCM)纯化得4a~4j。
4a: 淡褐色黏稠液体,收率78%;1H NMRδ: 8.40(d,J=6.5 Hz, 1H), 7.80(d,J=8.4 Hz, 2H), 6.44(d,J=6.3 Hz, 2H), 5.05~5.01(m, 1H), 4.30(t,J=7.8 Hz, 2H), 3.97 (t,J=6.1 Hz, 2H), 2.43(s, 3H), 1.99(s, 3H);13C NMRδ: 197.02, 163.56, 156.12, 132.42, 130.39, 128.62, 111.87, 59.12, 40.29, 28.54, 26.03。
4b: 黄色黏稠液体,收率84%;1H NMRδ: 8.42(d,J=6.3 Hz, 1H), 8.09(d,J=5.8 Hz, 2H), 7.79(d,J=8.4 Hz, 2H), 7.32~7.21(m, 3H), 6.45(d,J=6.4 Hz, 2H), 5.05~5.00(m, 1H), 4.31(t,J=7.4 Hz, 2H), 3.97~3.90(m, 2H), 2.45(s, 3H);13C NMRδ:196.88, 164.37, 156.08, 142.49, 132.26, 131.82, 130.36, 127.33, 126.74, 111.20, 59.46, 40.34, 26.01。
4c: 淡黄色固体,收率72%; m.p 149~151 ℃;1H NMRδ: 8.42(d,J=6.4 Hz, 1H), 8.28(s, 1H), 8.22(d,J=6.5 Hz, 2H), 7.80(d,J=8.3 Hz, 2H), 7.31~7.23(m, 1H), 6.48(d,J=6.1 Hz, 2H), 5.09~5.03(m, 1H), 4.31(t,J=7.8 Hz, 2H), 4.01(t,J=6.6 Hz, 2H), 2.48(s, 3H);13C NMRδ: 196.46, 166.34, 149.12, 134.95, 133.06, 132.12, 131.96, 130.34, 128.59, 127.81, 109.96, 106.58, 58.21, 40.98, 39.02, 29.67, 26.03。
4d: 淡黄色固体,收率82%; m.p.148~149 ℃;1H NMRδ: 8.46(d,J=6.3 Hz, 1H), 8.12(d,J=8.3 Hz, 2H), 7.31(d,J=5.8 Hz, 2H), 7.22~7.11(m, 2H), 6.34(d,J=6.8 Hz, 2H), 5.06~5.00(m, 1H), 4.36(t,J=7.8 Hz, 2H), 3.94 (t,J=6.4 Hz, 2H), 2.46(s, 3H);13C NMRδ: 196.47, 166.85, 154.23, 153.75, 135.19, 133.00, 132.04, 131.62, 130.90, 129.61, 126.84, 110.07, 106.56, 58.47, 40.73, 38.97, 26.02。
4e: 黄褐色黏稠液体,收率80%;1H NMRδ: 8.40(d,J=6.4 Hz, 1H), 8.11(d,J=6.0 Hz, 2H), 7.81(d,J=7.8 Hz, 2H), 7.21(d,J=5.4 Hz, 1H), 7.20~7.11(m, 2H), 6.46(d,J=6.3 Hz, 2H), 5.06~5.00(m, 1H), 4.32(t,J=7.5 Hz, 2H), 3.96~3.88 (m, 2H), 2.91(s, 2H), 2.47(s, 3H);13C NMRδ: 196.59, 164.87, 156.22, 141.61, 132.20, 131.91, 131.44, 130.05, 126.84, 126.72, 110.99, 60.01, 40.56, 39.00, 30.82, 26.04。
4f: 淡红色黏稠液体,收率78%;1H NMRδ: 8.71(d,J=6.4 Hz, 1H), 7.78(d,J=8.7 Hz, 2H), 6.42(d,J=6.4 Hz, 2H), 5.06~5.00(m, 1H), 4.30(t,J=7.8 Hz, 2H), 3.96~3.90(m, 2H), 3.01(s, 3H), 2.47(s, 3H);13C NMRδ:197.10, 155.77, 132.39, 131.02, 128.12, 110.75, 58.90, 47.23, 40.37, 26.02; HR-MS (ESI-TOF)m/z: Calcd for C12H16N2O3SNa {[M+Na]+} 291.077 9, found 291.078 1。
4g: 黄色黏稠状,收率72%;1H NMRδ: 8.70(d,J=6.4 Hz, 1H), 7.75(d,J=8.3 Hz, 2H), 7.24~7.15(m, 4H), 6.45(d,J=6.4 Hz, 2H), 5.05~4.99(m, 1H), 4.32(t,J=7.4 Hz, 2H), 3.99~3.88 (m, 2H), 2.48(s, 3H), 2.17(s, 3H);13C NMRδ: 196.79, 164.12, 156.00, 142.11, 132.20, 130.98, 130.26, 126.81, 126.02, 110.68, 59.42, 40.52, 26.00, 20.32。
4h: 黄褐色固体,收率64%, m.p.158~160 ℃;1H NMRδ: 8.72(d,J=6.4 Hz, 1H), 8.01(d,J=5.4 Hz, 2H), 7.72(d,J=8.4 Hz, 2H), 7.29~7.18(m, 2H), 6.47(d,J=6.5 Hz, 2H), 5.07~5.00(m, 1H), 4.30(t,J=7.4 Hz, 2H), 3.86 (t,J=7.4 Hz, 2H), 2.47(s, 3H);13C NMRδ:196.76, 164.41, 156.38, 142.27, 141.77, 133.42, 131.82, 130.05, 127.71, 124.68, 111.09, 59.37, 40.31, 26.01; HR-MS (ESI-TOF)m/z: Calcd for C17H17N2O3SFNa {[M+Na]+} 371.084 2, found 371.084 1。
4i: 黄褐色黏稠状液体,收率81%;1H NMRδ: 8.71(d,J=6.4 Hz, 1H), 7.74(d,J=6.4 Hz, 2H), 7.25~7.14(m, 5H), 6.46(d,J=6.4 Hz, 2H), 5.06~4.99(m, 1H), 4.35~4.30(m, 2H), 3.98~3.89(m, 2H), 2.47(s, 3H);13C NMRδ: 196.42, 164.62, 155.54, 142.130, 132.27, 131.41, 130.07, 130.26, 126.81, 126.72, 124.26, 120.34, 114.21, 110.52, 59.42, 40.54, 26.03。
4j: 红褐色固体,收率58%, m.p.163~165 ℃;1H NMRδ: 8.70(d,J=6.4 Hz, 1H), 7.96(d,J=8.4 Hz, 2H), 7.75(d,J=8.7 Hz, 2H), 7.22~7.11(m, 2H), 6.47(d,J=6.4 Hz, 2H), 5.05~4.98(m, 1H), 4.33~4.29(m, 2H), 3.98~3.87(m, 2H), 2.46(s, 3H);13C NMRδ:197.01, 165.53, 162.14, 144.58, 132.46, 130.91, 129.03, 128.59, 126.10, 125.29, 111.18, 59.61, 40.48, 26.02。
1.3细胞毒活性测试
以顺铂(DDP)为对阳性对照,以A549为待测细胞株,采用MTT法[10]测定4的体外细胞毒活性。取对数生长期的肿瘤细胞(5×104个·mL-1)接种于96孔板中,培养过夜后加入4,设置6个浓度梯度,每个浓度3个复孔。作用48 h后,每孔加入 5 mg·mL-1的MTT 20 μL,继续培养4 h,于3 000 rpm离心10 min,吸弃培养液,加入 DMSO 150 μL终止反应,于振荡器振荡10 min,用酶标仪测定波长570 nm处吸光度,并计算IC50,结果见表1。
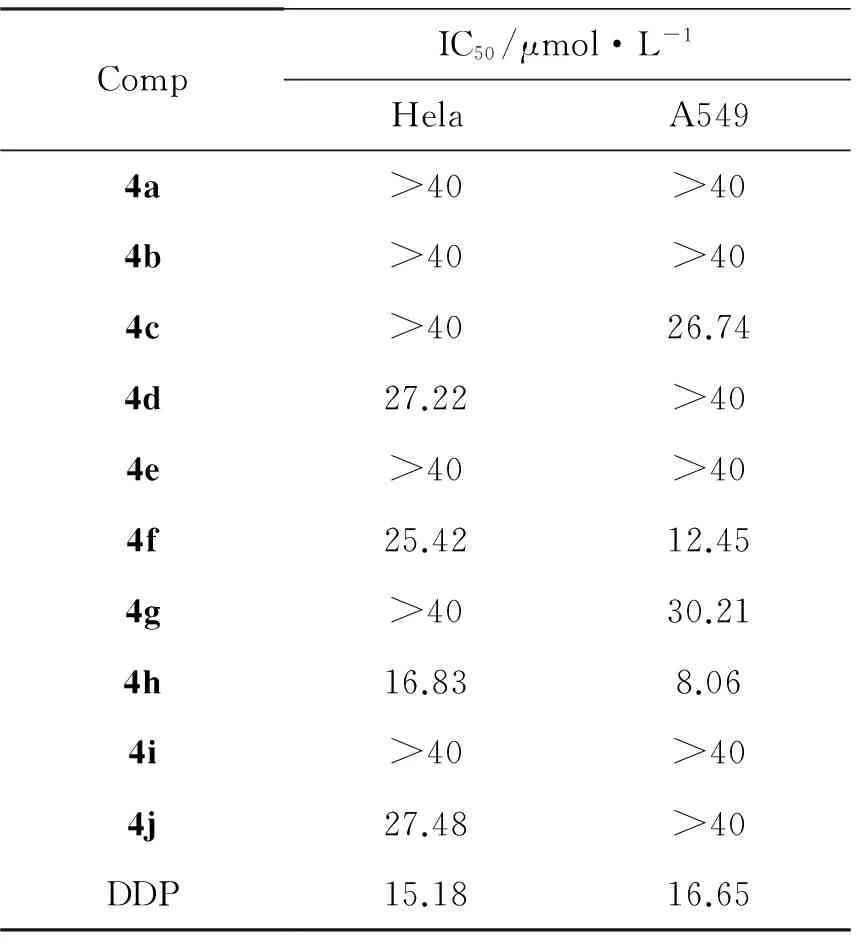
表1 4的体外细胞毒活性
2 结果与讨论
2.1合成
文献报道含氮杂环与4-氟苯乙酮的取代反应[11-12]大多采用柱层析色谱分离纯化得到目标产物。在1的合成中,我们对实验操作进行了优化。反应后直接加水使粗产物从水溶液中析出,且固体经20%乙醇水溶液洗涤,可除去粗产物中的杂质。通过此方法得到的化合物纯度较高,可直接进行下一步反应。
2.2体外细胞毒性
4的体外细胞毒性见表1。由表1可见,4对两种肿瘤细胞株的敏感度一般。4c和4d分别对细胞株A549和Hela表现出一般的抑制活性;磺酰胺衍生物对肿瘤细胞株的体外细胞毒活性较优于酰胺类化合物,尤其是4h,对A549和Hela的抑制活性与顺铂相当(IC50值分别为16.83 μmol·L-1和8.06 μmol·L-1),这为深入开展此类化合物的构效关系研究提供了参考依据。
以3-羟基氮杂环丁烷盐酸盐为原料,经5步反应合成制得10个3-氨基氮杂环丁烷衍生物,部分目标化合物对肿瘤细胞株A549和Hela表现出较好的体外抑制活性。为了研究不同取代基对分子活性的影响,更多衍生物的合成及活性研究正在进行中。
[1]Melloni P, Torre A D, Meroni M,etal. Azetidine derivatives of tricyclic antidepressant agents[J].J Med Chem,1979,22(2):183-191.
[2]Kozikowski A P, Tuckmantel W, Liao Y,etal. Synthesis and metabotropic receptor activity of the novel rigidified glutamate analogs (+)- and (-)-trans-azetidine-2,4-dicarboxylic acid and theirN-methyl derivatives[J].J Med Chem,1993,36(18):2706- 2708.
[3]张羲,崔杨. 两种氮杂环丁烷类药物中间体的合成[J].合成化学,2009,17(1):115-117.
[4]王贤洵,周国川. 1-苄氧羰基-3-叔丁氧羰酰氨基氮杂环丁烷的合成工艺改进[J].合成化学,2009,17(2):260-261.
[5]Sajjadi Z, Lubell W D. Amino acid-azetidine chimeras:Synthesis of enantiopure 3-substituted azetidine-2-carboxylic acids[J].The Journal of Peptide Research,2005,65(2):298-310.
[6]Sun Y Y, Cao Z, Gou S H. Novel oxaliplatin derivatives with 1-(substituted benzyl) azetidine-3,3- dicarboxylate anions.Synthesis,cytotoxicity,and interaction with DNA[J].Chemistry & Biodiversity,2014,11(1):115-125.
[7]Jones B, Proud M, Sridharan V. Synthesis of oxetane/azetidine containing spirocyclesviathe 1,3-dipolar cycloaddition reaction[J].Tetrahedron Letters,2016,57(25):2811-2813.
[8]Lawande P P, Sontakke V A, Nair R J,etal. Synthesis of polyhydroxylated azetidine iminosugars and 3-hydroxy-N-methylazetidine-2-carboxylic acid from d-glucose[J].Tetrahedron,2015,71(31):5085-5090.
[9]毛泽伟, 张梦迪, 饶高雄. 新型3-氨基氮杂环丁烷衍生物的合成[J].合成化学,2015,23(10):938-940.
[10]Ge Y, Cheng R, Chen Z,etal. Cryp to tanshinone induces cell cycle arrest and apoptosis of multidrug resistant human chronic my eloidleukemia cells by inhibiting the activity of eukaryotic initiation factor 4E[J].Mol Cell Biochem,2012,368:17-25.
[11]Mao Z W, Zheng X, Qi Y,etal. Synthesis and biological evaluation of novel hybrid compounds between chalcone and piperazine as potential antitumor agents[J].RSC Adv,2016,6:7723-7727.
[12]Mao Z W, Zheng X, Lin Y P,etal. Design, synthesis and anticancer activity of novel hybrid compounds between benzofuran andN-aryl piperazine[J].Bioorganic & Medicinal Chemistry Letters,2016,26:3421-3424.
Synthesis and Cytotoxic Activities of Azetidine Derivatives
ZHAO Shu-yan1,ZUO Ai-xue2,MAO Ze-wei2*
(1. Chengde Nursing Vocational College, Chengde 067000, China; 2. School of Traditional Chinese Medicine,Yunnan University of Traditional Chinese Medicine, Kunming 650500, China)
4-(3-aminoazetidine-1-yl)acetophenone(3)was synthesized by substitution and functional transformation using 3-hydroxyazetidine hydrochloride as starting material. Ten 3-aminoazetidine derivatives(4a~4j) were synthesized by derivatization of 3. Among them, 4f~4j were novel compounds. The structures were characterized by1H NMR,13C NMR and HR-MS(ESI-TOF). The preliminary cytotoxic activities were studiedinvitroagainst a panel of human tumor cell lines (Hela, A549) by the MTT assay. The result indicated that compound 4h was found to be the most potent compound against A549(IC50=8.06 μmol·L-1).
3-hydroxyazetidine hydrochloride; azetidine; derivative; synthesis; antineoplasmic activity
2016-07-04
云南省应用基础研究计划资助项目(2015FB155)
赵淑艳(1965-),女,满族,河北秦皇岛人,副教授,主要从事有机化学的研究。 E-mail: 421650043@qq.com
毛泽伟,博士, E-mail: ydmason@163.com
O625.42
ADOI: 10.15952/j.cnki.cjsc.1005-1511.2016.09.16168
