尾加压素Ⅱ诱导巨噬细胞分泌巨噬细胞集落刺激因子
2016-10-25路丹丁文惠彭芬李军赵静叶小巾
路丹 丁文惠 彭芬 李军 赵静 叶小巾
·基础研究·
尾加压素Ⅱ诱导巨噬细胞分泌巨噬细胞集落刺激因子
路丹丁文惠彭芬李军赵静叶小巾
目的探索尾加压素Ⅱ(UⅡ)能否促进小鼠单核巨噬细胞系RAW264.7细胞分泌巨噬细胞集落刺激因子(M-CSF),并探讨相关的信号机制。方法培养小鼠单核巨噬细胞系RAW264.7细胞,以Real-time PCR法和ELISA法分别检测细胞的M-CSF mRNA和蛋白表达水平,用细胞免疫印迹法检测p38MAPK和ERK的磷酸化水平。结果UⅡ呈浓度依赖性和时间依赖性促进RAW264.7细胞中M-CSF mRNA和蛋白水平的表达,最大效应时间均为刺激12 h,M-CSF mRNA水平是对照组水平的2.73倍,蛋白表达水平显著高于对照组水平[(50.04±4.28)pg/ml比(20.39±2.75) pg/ml,P<0.01],是对照组水平的2.45倍;最大效应浓度均为10-8mol/L和10-7mol/L,以10-8mol/L UⅡ刺激12 h,M-CSF的mRNA和蛋白表达水平较对照组分别增加了97.3%和80.8%。采用UⅡ受体(UT)阻断剂Urantide、ERK阻断剂PD98059和p38MAPK阻断剂SB203580分别预处理细胞后,M-CSF蛋白水平较UⅡ组显著降低,分别为[(45±4.04) pg/ml比(57.47±2.93)pg/ml]、[(42.13±4.28) pg/ml比(57.47±2.93)pg/ml]和[(44±5.34) pg/ml比(57.47±2.93)pg/ml],差异均有统计学意义(均P<0.05)。10-8mol/L UⅡ刺激细胞3 min显著促进ERK和p38MAPK蛋白磷酸化,5 min时ERK磷酸化水平达到峰值,至15 min仍持续高值,而p38MAPK在15 min时磷酸化达到峰值;30 min时,两者的磷酸化水平均减弱。结论UⅡ可通过与受体结合活化ERK和p38MAPK信号通路促进RAW264.7细胞中M-CSF的表达。
巨噬细胞集落刺激因子;炎症;尾加压素Ⅱ
尾加压素Ⅱ(urotensin Ⅱ, UⅡ)是一种重要的血管活性肽,自1999年发现UⅡ在人冠状动脉粥样硬化斑块中强表达以来,UⅡ在动脉粥样硬化中的作用备受关注[1]。本研究组前期研究显示,严重冠状动脉疾病患者血浆UⅡ水平升高,与冠心病的严重程度密切相关[2],载脂蛋白E缺陷鼠(ApoE-/-)第18、28和38周UⅡ受体(UT)表达逐渐增加[3]。另有研究报道UⅡ慢性灌注增加ApoE-/-鼠泡沫细胞和动脉粥样硬化斑块形成[4]。体外研究发现,UⅡ可促进多种炎症因子、血管生成因子的分泌,参与了动脉粥样硬化病理过程的多个环节[5-9]。本研究组前期研究发现UⅡ可通过p42/44丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)通路促进人脐静脉内皮细胞(human umbilical vein endothelial cells,HUVECs)增殖和抗凋亡,促进单核巨噬细胞分泌单核细胞趋化蛋白1(monocyte chemotactic protein 1,MCP-1)[5-6]。也有研究发现UⅡ可促进HUVECs增殖和分泌白介素8(IL-8),促进平滑肌细胞增殖和单核细胞趋化[7-9]。
巨噬细胞集落刺激因子(M-CSF)由受损的内皮细胞、平滑肌细胞及单核巨噬细胞分泌,在调节单核巨噬细胞的存活、分化、增殖和趋化上起着十分重要的作用。尽管UⅡ和M-CSF均可调节单核巨噬细胞的多种效应,但UⅡ能否促进巨噬细胞分泌M-CSF尚不明确。因此,本研究探索UⅡ能否促进小鼠单核巨噬细胞系RAW264.7细胞分泌M-CSF,并探讨相关信号机制。
1 材料与方法
1.1材料与试剂
小鼠单核巨噬细胞系RAW264.7细胞购自协和医学院细胞中心。DMEM培养基、Gibco胎牛血清(FBS)、TRIzol试剂、高保真cDNA逆转录试剂盒、SYBR®Select Master Mix均购自美国Life Technologies公司,小鼠M-CSF ELISA试剂盒购自美国Abcam公司,小鼠UⅡ购自美国Phoenix公司,UT阻断剂Urantide购自美国Peptides公司;p38MAPK通路阻断剂SB203580、细胞外信号调节激酶(Extra-cellular signal regulated kinase,ERK)通路阻断剂PD98059均购自美国Selleckchem公司。Real-time PCR引物由上海生工生物工程股份有限公司合成,序列如下:M-CSF F,5′-ACCGAGAGGCTCCAGGAA CT-3′;M-CSF R,5′-GTTCGGACACAGGCCTTGTT- 3′;β-actin F,5′-GGCCAACCGTGAAAAGATGA-3′;β-actin R, 5′- CACAGCCTGGATGGCTACGT-3′。磷酸化ERK和磷酸化p38 MAPK抗体、总ERK和总p38 MAPK抗体均购自美国CST公司。化学发光液购自美国Millipore公司。
1.2细胞培养和实验设计
常规紫外照射消毒超净台,在超净台内配含10% FBS的高糖DMEM培养基。将细胞培养在5%CO2的37℃孵箱。细胞呈圆形,汇合至80%时用培养基轻轻吹打下来,以1∶4~1∶6比例传代。取对数期细胞做实验。
将RAW261.7细胞接种于6孔板中,每孔约3×105个细胞,待细胞铺满皿底60%时,改用含1% FBS培养液同步化12 h后,对细胞分别进行如下干预:(1)分别用10-8mol/L UⅡ刺激0、 2、4、8、12、24和36 h,提取细胞总RNA,收集培养液上清于-80℃冰箱保存,以后做ELISA;(2)用不同浓度的UⅡ(10-10~10-7mol/L)刺激细胞12 h,提取细胞总RNA,收集培养液上清于-80℃冰箱保存,以后做ELISA;(3)用UT阻断剂Urantide(10-6mol/L)、ERK阻断剂PD98059(10-6mol/L)和p38 MAPK阻断剂SB203580(10-6mol/L)预处理细胞1 h后,再用10-8mol/L UⅡ刺激12 h,离心后提取细胞总RNA,收取上清于-80℃冰箱保存,以后做ELISA。
1.3细胞总RNA提取
Trizol法提取各组细胞的总RNA,紫外分光光度仪测定总RNA浓度和纯度, 在260 nm和280 nm波长下的光密度比值均为1.6~1.8。
1.4逆转录和Real-time PCR
按照高保真cDNA逆转录试剂盒说明,取各组细胞总RNA样本1 μg,加入逆转录酶1 μl,随机引物2 μl,10×RT缓冲液2 μl,25×dNTP混合物0.8 μl,总体积20 μl,逆转录合成cDNA。反应条件为25℃,10 min;37℃,120 min,85℃,5 min,温度降至4℃时取出cDNA进行Real-time PCR。
取cDNA产物1 μl做模板,将上游、下游引物稀释至2 μmol/L,采用非特异性染料(SYBR GreenⅠ)法进行Real-time PCR。20 μl体系包括cDNA 1 μl、上游引物2 μl(终浓度200 nmol/L)、下游引物2 μl(终浓度200 nmol/L)、dH2O 5 μl和Mix 10 μl。在ABI 7300 Real-time PCR仪上采用2步法进行反应。循环参数为变性温度95℃,10 min;退火温度95℃,15 s;延伸温度60℃,1 min,共40个循环,4℃保存。同时增加溶解曲线观察扩增情况。反应结束后从仪器上读取CT值,以同一模板中目的基因和管家基因β-actin的相对浓度的比值作为目的基因相对转录水平,采用2-ΔΔCT法在各实验组中比较。
1.5ELISA
收集各组细胞培养基上清,以1000 r/min离心10 min,取上清分装置于1.5 ml EP管中于-80℃冰箱保存备用。实验操作按照试剂盒说明书进行。
1.6免疫印迹检测
将各组细胞弃上清,以PBS冲洗3遍,加入蛋白裂解液提取总蛋白,用蛋白质定量试剂盒(BCA法)测定蛋白浓度, 95℃,5 min。配10% SDS-PAGE凝胶,上样量约20 μg进行电泳,浓缩胶80 V,30 min,分离胶100 V,1 h;200 mA 转膜1.5 h;牛血清蛋白溶液(5%BSA)封闭1 h;一抗(p-ERK、p-p38MAPK均1∶1000)4℃过夜;TBST洗涤3次,二抗(1∶8000)孵育1 h;TBST洗涤3次,发光。膜再生液洗涤30 min,重新封闭1 h,孵总蛋白(ERK、p38MAPK均1∶1000)过夜;TBST洗涤3次,二抗(1∶8000)孵育1 h;TBST洗涤3次,发光。Image J分析灰度值,计算磷酸化蛋白/总蛋白。
1.7统计学分析
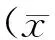
2 结果
2.1用Real-time PCR测定M-CSF的mRNA水平情况
Real-time PCR测定UⅡ(10-8mol/L)不同时间刺激RAW264.7细胞发现,UⅡ刺激4 h能引起M-CSF mRNA水平增加(P<0.05),较基线提高了约64.2%,刺激12 h达到最大值(P<0.01),约为对照组的2.73倍,刺激36 h仍持续在较高水平(P<0.01)(图1 A);不同浓度的UⅡ(10-9~10-7mol/L)刺激RAW264.7细胞12 h均能显著促进M-CSF mRNA表达(均P<0.01),浓度为10-8mol/L和10-7mol/L时达到最大值,10-8mol/L时较对照组约增加了97.3%(P<0.01,图1 B)。因此,UⅡ呈浓度和时间依赖性地促进RAW264.7细胞中M-CSF mRNA水平的表达。
2.2以ELISA法测定M-CSF蛋白的分泌情况
用ELISA法测定不同时间UⅡ(10-8mol/L)刺激RAW264.7细胞发现,刺激4 h后M-CSF蛋白分泌量较对照组显著增加[(29.64±3.55) pg/ml比(20.39±2.75) pg/ml,P<0.05],增加了45.4%;刺激12 h后,M-CSF蛋白水平达到最大值[(50.04±4.28)pg/ml比(20.39±2.75) pg/ml,P<0.01],约为对照组的2.45倍;刺激36 h仍持续在较高水平(P<0.01)(图2 A)。不同浓度的UⅡ(10-9、10-8、10-7mol/L)刺激12 h后,M-CSF的分泌量较对照组显著增加[(39.02±2.66)pg/ml比(25.85±2.49)pg/ml、(46.74±2.77) pg/ml比(25.85±2.49)pg/ml、(52.11±2.78) pg/ml比(25.85±2.49)pg/ml],差异均有统计学意义(均P<0.01),浓度为10-8mol/L和10-7mol/L时达到峰值,10-8mol/L时较对照组约增加了80.8%(图2 B)。因此,UⅡ呈时间和浓度依赖性地促进RAW264.7细胞分泌M-CSF蛋白。
2.3p38MAPK通路和ERK通路参与M-CSF蛋白分泌情况
为了探索p38MAPK通路和ERK通路是否参与了UⅡ诱导的M-CSF释放,用UT阻断剂Urantide、ERK阻断剂PD98059和p38MAPK阻断剂SB203580分别预处理细胞1 h,然后用UⅡ(10-8mol/L)刺激12 h, 检测M-CSF表达水平。结果显示,Urantide、PD98059和SB203580预处理细胞后较UⅡ组M-CSF的mRNA水平分别降低了54.3%、51.2%和49.7%。UⅡ组M-CSF水平较对照组显著升高[(57.47±2.93) pg/ml比(31.38±4.22) pg/ml,P<0.01,图3A]。Urantide、PD98059和SB203580预处理细胞后M-CSF的蛋白水平较UⅡ组显著降低[(45±4.04) pg/ml比(57.47±2.93) pg/ml、(42.13±4.28) pg/ml比(57.47±2.93) pg/ml、(44±5.34) pg/ml比(57.47±2.93) pg/ml],分别降低了47.8%、58.8%和51.6%,差异均有统计学意义(均P<0.05,图3B)。表明UT、ERK通路和p38MAPK通路均参与了UⅡ诱导的M-CSF分泌。
2.4用细胞免疫印迹检测p38MAPK和ERK的磷酸化情况
以10-8mol/L UⅡ刺激RAW264.7细胞0、3、5、15和30 min,发现UⅡ刺激3 min,p38MAPK、ERK显著磷酸化,5 min时ERK磷酸化水平达到峰值,至15 min仍持续高值,而p38MAPK在15 min时磷酸化达到峰值;30 min时,两者的磷酸化水平均减弱(图4)。

图1 用Real-time PCR测定RAW264.7细胞中M-CSF的mRNA表达 M-CSF,巨噬细胞集落刺激因子;UⅡ,尾加压素Ⅱ;a,与对照组比较,P<0.05;b,与对照组相比,P<0.01(n=3)
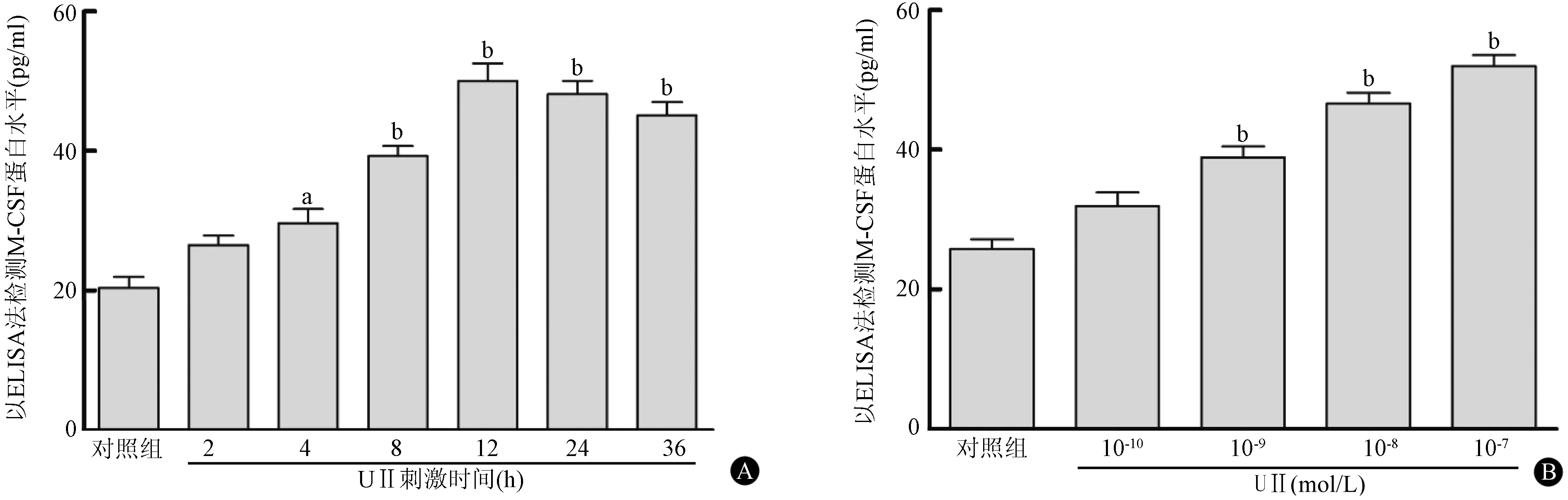
图2 ELISA法测定RAW264.7细胞中M-CSF的蛋白分泌 M-CSF,巨噬细胞集落刺激因子;UⅡ,尾加压素Ⅱ;a,与对照组相比,P<0.05;b,与对照组相比,P<0.01(n=3)

图3 ERK通路和p38MAPK通路参与了UⅡ诱导的M-CSF分泌 UA,尾加压素Ⅱ受体拮抗剂Urantide,10 μmol/L; PD,ERK通路阻断剂PD98059,10 μmol/L;SB,p38MAPK通路阻断剂SB203580, 10 μmol/L;UⅡ,尾加压素Ⅱ;M-CSF,巨噬细胞集落刺激因子;ERK,细胞外信号调节激酶;p38MAPK,p38丝裂原活化蛋白激酶;a,与对照组相比,P<0.05;b,与对照组相比,P<0.01;c,与UⅡ组相比,P<0.05(n=3)
3 讨论
单核巨噬细胞是动脉粥样硬化病理过程中最重要的炎症细胞,在动脉粥样硬化的各个环节——从泡沫细胞的形成到斑块进展、斑块不稳定及斑块破裂都发挥了重要作用[10]。抑制单核细胞进入血管壁形成巨噬细胞可能减缓动脉粥样硬化的进展[11-12]。单核细胞和巨噬细胞还是表达UT最多的细胞[7],UⅡ在泡沫细胞丰富的区域强表达,提示UⅡ/UT可能在单核巨噬细胞的效应中发挥重要作用[1]。
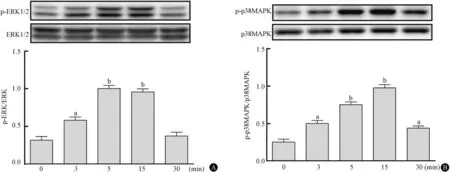
图4 UⅡ促进ERK和p38MAPK蛋白磷酸化 UⅡ,尾加压素Ⅱ;ERK,细胞外信号调节激酶;p38MAPK,p38丝裂原活化蛋白激酶;a,与基线组相比,P<0.05;b,与基线组相比,P<0.01(n=3)
M-CSF与冠心病密切相关。M-CSF在稳定型和不稳定型心绞痛患者中均升高[13-14],是心绞痛患者远期冠心病事件的独立预测因子[13]。体外试验也证实M-CSF是调节单核巨噬细胞系存活、生长、分化和趋化的细胞因子[15],M-CSF还可刺激局部内皮细胞和巨噬细胞产生更多的M-CSF[16]。M-CSF刺激巨噬细胞后立即引起细胞极化,细胞骨架重组并迁移到M-CSF所在部位。本研究结果发现,UⅡ可呈时间和浓度依赖性地促进M-CSF在RAW264.7细胞中mRNA的表达和蛋白分泌。本研究组前期工作还发现UⅡ可促进MCP-1的分泌[5],表明UⅡ是一种致炎和致动脉粥样硬化因子,可能通过促进单核巨噬细胞分泌M-CSF促进细胞存活,促进局部更多M-CSF生成,通过M-CSF和MCP-1趋化更多的单核巨噬细胞到炎症部位,从而加重局部的炎症反应,促进和维持动脉粥样硬化形成。
本研究发现10-8mol/L UⅡ刺激12 h时M-CSF的表达量达到较高水平。有文献报道20 nmol/L UⅡ刺激人冠状动脉内皮细胞6 h,组织因子水平显著增加,刺激12 h还可显著增加细胞黏附因子1和血管细胞黏附因子1的表达[17]。 30 nmol/L UⅡ刺激HUVECs 24 h,IL-8表达水平较高[8]。本研究组前期工作也发现10-8mol/L UⅡ刺激大鼠血管外膜成纤维细胞24 h, 血管内皮生长因子、白三烯C4蛋白水平达到高峰[18-19]。这些结果均表明,UⅡ刺激较短时间可促进血管壁细胞合成细胞黏附分子,促进单核巨噬细胞分泌趋化因子,从而促进炎症细胞黏附到血管壁和促进炎症细胞趋化到炎症部位;刺激较长时间则促进炎症因子的分泌。即UⅡ的效应可能与UⅡ刺激的细胞类型、所检测因子的性质和功能有关。
UⅡ诱导M-CSF在RAW264.7细胞中分泌的机制与UⅡ诱导IL-8在HUVECs、白三烯C4在大鼠胸主动脉外膜成纤维细胞中分泌的机制相似[8, 20]。UⅡ可通过与受体结合后活化p38MAPK通路和ERK通路促进HUVECs合成IL-8[8]、促进大鼠胸主动脉外膜成纤维细胞分泌白三烯C4。M-CSF的生成也可受p38MAPK通路和ERK通路调控[21]。本研究发现UⅡ通过与受体结合,活化p38MAPK通路和ERK通路促进单核巨噬细胞分泌M-CSF,这些通路可能在UⅡ介导的单核巨噬细胞的各种效应中发挥作用。
综上所述,UⅡ可通过活化UT,激活p38MAPK通路和ERK信号转导通路促进M-CSF在RAW264.7细胞中mRNA和蛋白水平的表达。本研究提供了新的UⅡ效应的视角,UⅡ可能促进巨噬细胞自分泌M-CSF,从而促进动脉粥样硬化的血管壁炎症反应。阻断UT可能抑制巨噬细胞分泌M-CSF,一定程度上延缓动脉粥样硬化进程。
[1] Ames RS, Sarau HM, Chambers JK, et al. Human urotensin-Ⅱ is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature, 1999,401(6750):282-286.
[2] 张丽芳, 陈莉, 丁文惠, 等. 血浆尾加压素Ⅱ浓度与冠状动脉病变程度的关系.中日友好医院学报, 2008,22(1):3-6.
[3] Wang ZJ, Shi LB, Xiong ZW, et al. Alteration of vascular urotensin Ⅱ receptor in mice with apolipoprotein E gene knockout. Peptides, 2006,27(4):858-863.
[4] Shiraishi Y, Watanabe T, Suguro T, et al. Chronic urotensin Ⅱ infusion enhances macrophage foam cell formation and atherosclerosis in apolipoprotein E-knockout mice. J Hypertens, 2008,26(10):1955-1965.
[5] 邸北冰, 丁文惠, 赵静, 等. 尾加压素Ⅱ诱导巨噬细胞表达单核细胞趋化蛋白1上调及机制研究. 中华老年心脑血管病杂志, 2011,13(10):925-928.
[6] Shi L, Ding W, Li D, et al. Proliferation and anti-apoptotic effects of human urotensin Ⅱ on human endothelial cells. Atherosclerosis, 2006,188(2):260-264.
[7] Segain JP, Rolli-Derkinderen M, Gervois N, et al. Urotensin Ⅱ is a new chemotactic factor for UT receptor-expressing monocytes. J Immunol, 2007,179(2):901-909.
[8] Lee CY, Tsai YT, Loh SH, et al. Urotensin Ⅱ induces interleukin 8 expression in human umbilical vein endothelial cells. PLoS One, 2014,9(2):e90278.
[9] Rodríguez-Moyano M, Díaz I, Dionisio N, et al. Urotensin-Ⅱ promotes vascular smooth muscle cell proliferation through store-operated calcium entry and EGFR transactivation. Cardiovasc Res, 2013,100(2):297-306.
[10] Laguna JC, Alegret M. Regulation of gene expression in atherosclerosis: insights from microarray studies in monocytes/macrophages. Pharmacogenomics, 2012,13(4):477-495.
[11] Moulton KS, Vakili K, Zurakowski D, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A, 2003,100(8):4736-4741.
[12] Jaipersad AS, Lip GY, Silverman S, et al. The role of monocytes in angiogenesis and atherosclerosis. J Am Coll Cardiol, 2014,63(1):1-11.
[13] Ikonomidis I, Lekakis J, Revela I, et al. Increased circulating C-reactive protein and macrophage-colony stimulating factor are complementary predictors of long-term outcome in patients with chronic coronary artery disease. Eur Heart J, 2005,26(16):1618-1624.
[14] Hojo Y, Ikeda U, Takahashi M, et al. Increased levels of monocyte-related cytokines in patients with unstable angina. Atherosclerosis, 2002,161(2):403-408.
[15] Thorpe R, Wadhwa M, Bird CR, et al. Detection and measurement of cytokines. Blood Rev, 1992,6(3):133-48.
[16] Roth P, Stanley ER. The biology of CSF-1 and its receptor. Curr Top Microbiol Immunol, 1992,181:141-167.
[17] Cirillo P, De Rosa S, Pacileo M, et al. Human urotensin Ⅱ induces tissue factor and cellular adhesion molecules expression in human coronary endothelial cells: an emerging role for urotensin Ⅱ in cardiovascular disease. J Thromb Haemost, 2008,6(5):726-736.
[18] Song N, Ding W, Chu S, et al. Urotensin Ⅱ stimulates vascular endothelial growth factor secretion from adventitial fibroblasts in synergy with angiotensin Ⅱ. Circ J, 2012,76(5):1267-1273.
[19] Dong X, Ye X, Song N, et al. Urotensin Ⅱ promotes the production of LTC4 in rat aortic adventitial fibroblasts through NF-kappaB-5-LO pathway by p38 MAPK and ERK activations. Heart Vessels, 2013,28(4):514-523.
[20] Djordjevic T, BelAiba RS, Bonello S, et al. Human urotensin Ⅱ is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler Thromb Vasc Biol, 2005,25(3):519-525.
[21] Guan SM, Zhang M, He JJ, et al. Mitogen-activated protein kinases and phosphatidylinositol 3-kinase are involved in Prevotella intermedia-induced proinflammatory cytokines expression in human periodontal ligament cells. Biochem Biophys Res Commun, 2009,386(3):471-476.
Urotensin Ⅱ induces macrophages to produce macrophage colony-stimulating factor
LU Dan, DING Wen-hui, PENG Fen, LI Jun, ZHAO Jing, YE Xiao-jin.
Department of Cardiology, Peking University, First Hospital, Beijing 100034, China Corresponding author: DING Wen-hui, Email:dwh_rd@126.com
ObjectiveTo investigate the role of urotensin Ⅱ in M-CSF secretion from RAW264.7 macrophages and the relevant signal mechanisms. MethodsThe RAW264.7 cell line was cultured. The mRNA expression and protein level of M-CSF were determined by real time polymerase chain reaction and Enzyme-linked immunosorbent assay, respectively. Western blot was applied to assay the phosphorylation status of ERK and p38MAPK. ResultsUⅡ increased the expression of M-CSF mRNA and protein in a concentration- and time-dependent manner with the peak at 12 h of treatment (P<0.01), when the gene and protein level of M-CSF was about 1.73 and 1.45 times more than the baseline level, respectively. The maximal effect was reached at 10-8mol/L and 10-7mol/L (bothP<0.01). Compared with the control group, the mRNA and protein expression of M-CSF increased by 97.3% and 80.8% after 12 h of UⅡ stimulation. The UⅡ effects were significantly inhibited by its receptor (UT) antagonist urantide, the SB203580 p38MAPK inhibitor and the PD98059 ERK inhibitor. 10-8mol/L UⅡ also enhanced ERK and p38MAPK phosphorylation in RAW264.7 macrophages. ConclusionsUrotensin Ⅱ promotes M-CSF release in RAW264.7 macrophages via p38MAPK and ERK signaling pathways.
Macrophage colony-stimulating factor;Inflammation;Urotension Ⅱ
10.3969/j.issn.1004-8812.2016.08.007
高等学校博士学科点专项科研基金(20120001120010)
100034北京,北京大学第一医院心内科
丁文惠,Email:dwh_rd@126.com
R363
2016-04-29)
