AMPK信号通路调控的自噬在七氟烷后处理保护大鼠心肌缺血再灌注损伤中的机制研究*
2016-10-18覃琴王琛乔世刚周迅
覃琴,王琛,乔世刚,周迅
(苏州大学附属第二医院麻醉科,苏州 215004)
1 引 言
缺血性心肌病具有高发病率和高死亡率的特点,是世界常见的死亡原因之一。有效及时地恢复冠状动脉血流是减少心肌梗死,提高患者存活率的有效途径。然而再灌注会导致心脏出现严重的心率失常、收缩舒张功能障碍以及心肌梗死范围增加等一系列并发症[1-2]。已证实吸入麻醉药预处理与后处理能减少心肌缺血再灌注(Ischemia/Reperfusion, I/R)损伤[3-4]。2003年Kemp BE首次报道腺苷酸活化蛋白激酶(AMP-activated protein kinase, AMPK)是哺乳动物体内一个复杂的异源性三聚体结构,一直被作为一个“细胞内燃料计”或“能量调节器”而被人们所关注。近来研究表明,AMPK的激活在调节机体氧化应激、KATP通道、细胞增值和死亡中均发挥重要作用[5-6],这些机制可能与AMPK激活后发挥I/R心肌保护作用有关。并且AMPK作为调节细胞内自噬/溶酶体途径的上游信号通路,一直以来都是人们关注的热点[7-9]。我们在前期研究的基础上,通过给予AMPK激活抑制剂Compound C,观察心肌损伤程度和检测p-AMPK/t-AMPK蛋白表达及其通路下游自噬相关蛋白表达变化,进一步探讨七氟烷后处理产生的心肌保护机制。
2 材料与方法
2.1 动物I/R模型建立及分组
健康雄性成年SD大鼠50只,质量270~320 g,SPF级,由昭衍(苏州)新药研究中心有限公司提供,许可证编号[scxk(苏)2014-0006]。所有的实验操作均经苏州大学实验动物使用及保护委员会许可。
50只大鼠建立在体心肌I/R模型,戊巴比妥钠50 mg/kg腹腔内注射麻醉大鼠后,于右侧颈内静脉及颈内动脉置入充满肝素的导管,用于静脉给药、动脉血气分析或监测动脉血压。气管切开并插入气管导管,连接ALC-V9动物呼吸机,行呼气末正压通气,吸入氧浓度33%,调节呼吸频率或潮气量,维持pH 7.35~7.45、PaCO2 25~40 mmHg、PaO2 90~150 mmHg (1mmHg=0.133KPa)。采用智能恒温控制仪维持大鼠体温在36 ℃~37 ℃。于大鼠第5肋间行左胸切开术,打开心包,6~0无损伤缝合线在左心耳下缘结扎左冠状动脉前降支(left anterior descending coronary artery,LAD),缝线末端穿入自制圈套管,平衡30 min。用止血钳夹紧圈套管以阻断LAD血供,若心外膜发绀苍白,心电图示一过性心律失常,ST段弓背向上抬高表示缺血模型成功;松开圈套管进行再灌注,可见心外膜重新充血证明再灌注成功[10],大鼠因再灌注期频发心律失常删除。
I/R模型建立成功后,随机将50大鼠分为5组:对照组(Sham组),不缺血维持5 h;I/R组,平衡30 min,缺血30 min,再灌注4 h;七氟烷后处理组(SP组),平衡30 min,缺血30 min,在缺血27 min时,给予大鼠吸入2.5%七氟醚(批号:H20130816,Maruishi Pharmaceutical公司,日本)3 min,再灌注开始后2 min继续吸入七氟醚后停止,共5 min;DMSO组,平衡30 min,缺血30 min,再灌注开始后静脉给予等量的Compound c溶剂二甲亚砜(DMSO),再灌注4 h; Compound c组(Com c组),平衡30 min,缺血30 min,再灌注开始后静脉给予AMPK抑制剂Compound c 250 g/kg[11],再灌注4 h。通过生物信号采集处理系统连续监测并记录各组大鼠平衡15、缺血15 min、再灌注1、2和4 h等时间点心率、平均动脉压(MAP)和心率-收缩压乘积(rate-pressure product, RPP)。
2.2 TTC法测定心肌梗死范围
大鼠心肌再灌注4 h时,再次阻断LAD,颈内静脉注射5%伊文思蓝1 ml使左心室正常区域蓝染,迅速取出心脏分离出左心室,用干冰速冻5 min,使用大鼠心脏切片器横断分割成6块2 mm厚的组织块。将左心室中蓝染的正常组织与未染色的左心室缺血区组织分离。采用TTC染色法,将心肌组织放入1% TTC中,置于37℃恒温水浴箱15 min,4%多聚甲醛过夜以固定组织。立式显微镜下将左心室分成正常(蓝染区域)、缺血未梗死区(TTC染色后红色区域)和缺血梗死区(TTC染色后灰白色区域)3部分,并分别用电子天平称重,计算缺血区心肌质量(缺血未梗死区心肌质量+梗死区心肌质量)占左心室心肌质量的百分比、缺血梗死区心肌质量占缺血区心肌质量的百分比,心肌梗死范围以梗死区心肌质量占缺血区心肌质量的百分比表示[10]。
2.3 Western Blot法测定p-AMPK/t-AMPK、LC3、P62蛋白表达
随机将30大鼠分为5组,每组6只。再灌注4 h迅速取下心脏,-80 ℃液氮冷冻保存。取约300 mg左心室心尖部组织,剪碎,置于匀浆器最底部,加入Western细胞裂解液,蛋白酶抑制剂(PMSF)和磷酸酶抑制剂(PST),低温下超声细胞破碎仪裂解3次。裂解后,低温下静置半小时,将裂解后的组织匀浆移至1.5 ml EP管中,将EP管放入离心机中,4℃下1 500 g离心30 min,取上清分装于1.5 ml离心管中,并于低温冰箱中保存。所有操作均必须在冰面进行。运用BCA蛋白浓度测定试剂盒测定提取的蛋白样品的浓度,并用裂解液将其调至相同蛋白浓度。在样品中加入上样缓冲液混匀,95 ℃煮5 min,使蛋白质充分变性,并于低温冰箱中冻存。BCA蛋白浓度测定试剂盒(批号:P0010S,碧云天生物技术研究所)测定蛋白样品的蛋白含量,并用裂解液调至相同浓度值,97 ℃金属浴持续加热5 min。每个样品取20 μg蛋白于12%聚丙烯酰胺凝胶(BIO-RAD公司,美国)分离蛋白,电泳完毕转至硝酸纤维素膜(Millipore公司,美国),5%脱脂牛奶封闭2 h,分别加入一抗p-AMPK(1∶1000,批号:2535,Cell Signaling公司,美国)、一抗LC3(1∶1000,批号:136F3,Santa Cruz公司,美国)、一抗P62(1∶1000,批号:8G10,Santa Cruz公司,美国)、内参GAPDH(1:1000,批号:AG019,碧云天生物技术研究所),4℃一抗孵育过夜。TBST溶液漂洗3遍后加入二抗摇床孵育2 h。采用ECL试剂盒(批号:34077,Thermo公司,美国)显色;最后进行增强化学发光反应,将PVDF膜放入发光液(免疫印迹化学发光试剂AB液,批号:0609*025,Merck公司,德国)中,移置暗室,曝光后自动洗片机显影,定影。采用Image软件测定条带灰度值,以目的条带灰度值与GAPDH灰度值的比值反映心肌组织p-AMPK/t-AMPK、LC3、P62蛋白表达水平。
2.4 统计学方法

3 结果
3.1 各组血流动力学指标比较
平衡15min,各组间心率、MAP和RPP比较差别无统计学意义(P>0.05)。在缺血期和再灌注期,除Sham组外,其余各组MAP和RPP较平衡期下降(P<0.05);与SP组比较,DMSO组差别均无统计学意义(P>0.05),I/R组和Com c组MAP和RPP下降(P<0.05)。各组再灌注期间大鼠死亡率无统计学意义(P>0.05)(见表1)。

表1 各组大鼠血流动力学变化的比较
注:与平衡期比较,P<0.05;与I/R组比较,P<0.05;与SP组比较,P<0.05
3.2 各组心肌梗死范围比较
各组心肌缺血区域所占比例比较无明显差异(P>0.05)。与I/R组(55.2±11.6%)比较,SP组(36.8±17.2%)心肌梗死范围减少(P<0.05);与SP组比较,Com c组(56.6 ± 13.2%)梗死范围增大(P<0.05);SP组和DMSO组比较差异无统计学意义(P>0.05)(见表2,图1)。
3.3 Western Blot检测结果
与I/R组比较,SP组和DMSO组p-AMPK/t-AMPK蛋白表达上调而LC3 Ⅱ/Ⅰ、P62蛋白表达下调(P<0.05);与SP组比较,Com c组p-AMPK/t-AMPK蛋白表达下调而LC3 Ⅱ/Ⅰ、P62蛋白表达上调(P<0.05)(见图2、图4)。
表2 各组缺血再灌注心肌各部分质量及比值

注:与Sham组比较,aP<0.05,bP<0.01;与I/R组比较,cP<0.05;与SP组比较,eP<0.05
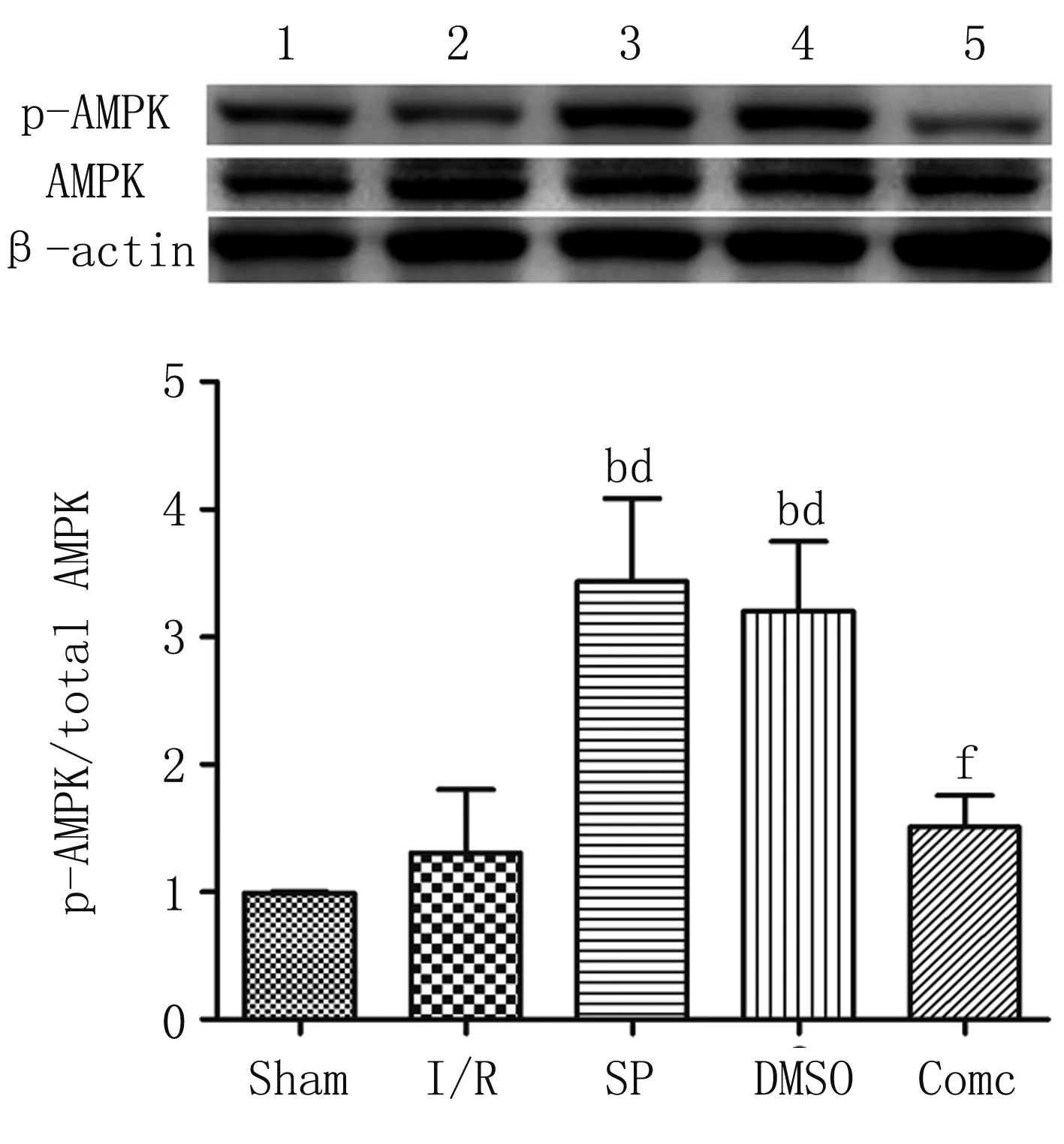
与Sham组比较,bP<0.01;与I/R组比较,dP<0.01;与SP组比较,fP<0.01

与Sham组比较,aP<0.05,bP<0.01;与I/R组比较,cP<0.05;与SP组比较,eP<0.05
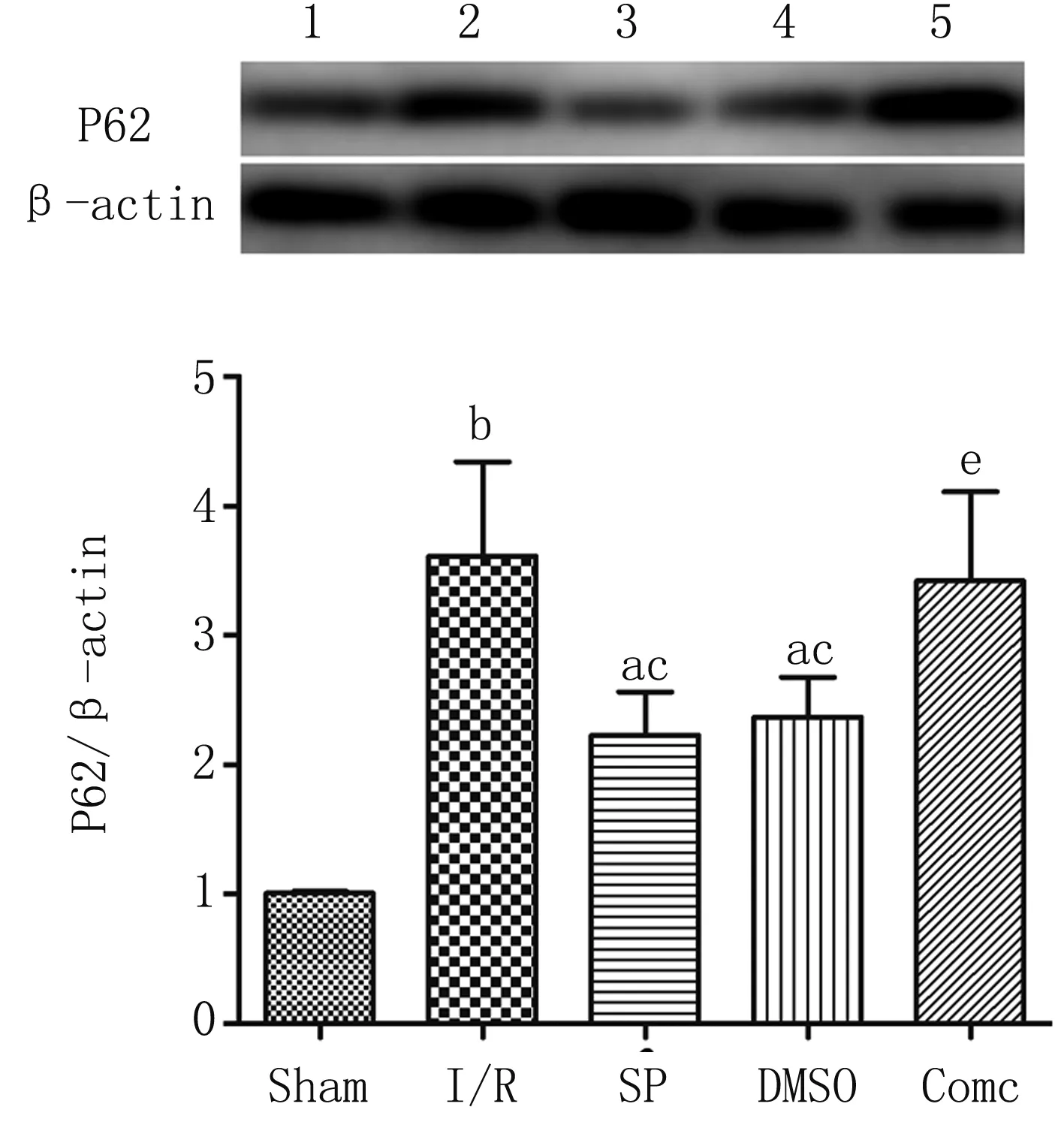
与Sham组比较,aP<0.05,bP<0.01;与I/R组比较,cP<0.05;与SP组比较,eP<0.05
4 讨论
AMPK是一个复杂的异源性三聚体丝氨酸/苏氨酸蛋白激酶,一个催化亚基α亚基,两个调节亚基β亚基和γ亚基。其α亚基172位苏氨酸的磷酸化结合后可以激活AMPK的活性。多种病理和生理状态下如缺血低氧、运动、AMP/ATP比例的升高以及促炎因子等,均可以使AMPK磷酸化激活[12-13]。激活的AMPK通过多种途径促进ATP的生成以及脂肪酸的氧化,因此AMPK一直被称为细胞内能量调节器,在心肌I/R损伤中,已有研究证明激活的AMPK对心肌具有保护作用[5-6],这与我们实验研究结果一致,七氟烷后处理组心肌梗死范围减少,具有心肌保护效应,同时 p-AMPK/t-AMPK蛋白表达上调;给予AMPK抑制剂Compound c后p-AMPK/t-AMPK蛋白表达受到抑制,心肌梗死范围增加,心肌保护效应消失。
自噬是细胞内溶酶体降解长寿蛋白质及损伤的细胞器从而维持胞内稳态平衡的一种生物过程[14]。只有完整的自噬流形成自噬溶酶体才能发挥细胞保护作用[15]。自噬在正常情况下表达水平相对较低,在某些应激的情况下可使其激活适应应激反应,发挥保护作用。然而,研究发现在死亡的细胞内存在着大量的自噬体[16],过量的自噬可以导致细胞的死亡(即Ⅱ型程序性死亡)。Kabeya等2000年研究发现LC3因能始终靶向定位于自噬体膜上直到与溶酶体溶合,是目前确认的唯一可信的自噬体标记物,LC3的水平在某种程度上反映了自噬小体的数量。P62是一种泛素结合蛋白,与蛋白质的泛素化密切相关,它参与多种细胞信号转导调控及自噬过程[17]。自噬过程中,P62与泛素化的蛋白质结合,再与定位于自噬小体内膜上的 LC3-II 蛋白形成复合物,一同在自噬溶酶体内降解。因此,出现自噬时,在细胞质中P62蛋白不断被降解;当自噬活性减弱、自噬功能缺陷时,P62蛋白会在细胞质中不断累积,P62是反映自噬活性的标记蛋白之一,其含量间接反映自噬小体清除水平[18]。
已有研究发现缺血和再灌注两个阶段中自噬是持续进行的,吞噬包裹所形成的自噬泡需要与溶酶体结合消化才能形成完整的自噬过程(即完整的自噬流),然而I/R破坏了其结合的过程,从而使得自噬泡大量的聚集最终导致自噬性死亡[9]。同时也有报道七氟烷后处理修复完整的自噬流,恢复再灌注期间溶酶体功能,发挥I/R心肌保护作用[19]。缺血期通过AMPK通路激活的自噬对心肌发挥保护作用,而再灌注时通过Beclin-1途径激活的自噬则发挥出相反的作用[8]。近期有研究报道通过激活AMPK信号通路,保护自噬泡与溶酶体结合即自噬流,从而抑制再灌注期间细胞内ROS的生成和细胞死亡[20]。这与我们研究相符,七氟烷后处理上调p-AMPK/t-AMPK蛋白的表达,说明AMPK信号通路参与了七氟烷后处理保护机制,给予AMPK抑制剂Compound c后此保护作用消失;同时七氟烷后处理降低再灌注后自噬体标志性蛋白LC3和溶酶体功能损伤标志性蛋白P62的表达,说明七氟烷后处理可以修复再灌注后自噬流的破坏,对在体心肌I/R损伤发挥保护作用。此外,比较SP组和I/R组的结果发现,给予AMPK的抑制剂Compound c增加了LC3和P62蛋白的表达,这表明AMPK信号通路对细胞内自噬流具有重要的调节作用。
综上所述,七氟烷后处理对大鼠在体心肌I/R损伤具有保护作用,其机制可能是通过激活AMPK信号通路,使AMPK磷酸化,从而保护了自噬流,促使自噬体与溶酶体结合,使得自噬发挥其消化降解代谢产物并循环利用的作用,对心肌I/R损伤产生保护作用,这对今后我们研究七氟烷后处理产生心肌保护作用的机制提供了新思路。
