基于罗丹明B和金纳米粒子荧光共振能量转移检测卡托普利
2016-10-16薛李慧王靖原孙雪花栾建明李晓江
田 锐, 薛李慧, 马 晨, 王靖原, 孙雪花, 栾建明, 李晓江
(1.延安大学化工学院,陕西延安 716000;2.延安市实验中学,陕西延安 716000)
卡托普利(Captopril)是第一代口服血管紧张素转换酶抑制剂,临床上广泛用于治疗各种高血压、冠心病等,然而当卡托普利用量过大时,会导致肾脏损害、循环系统衰竭等症状[1,2],因此,建立准确、灵敏测定卡托普利含量的方法对指导临床用药有着重要的意义。目前,测定卡托普利的方法主要有高效液相色谱法[3 - 5]、化学发光法[6]、光度法[7]、原子吸收法[8]、流动注射分析法[9]和电泳法[10]等。但这些方法或需要昂贵的仪器、或操作繁琐,制约了它们的应用。
近年来,金纳米粒子(Gold Nanoparticles,AuNPs)因其制备简单、生物兼容性好、催化活性高,尤其是良好的光学性质而得到广泛应用[11]。基于分散态AuNPs对邻近荧光基团(如荧光染料)荧光猝灭的荧光分析法被广泛用于DNA、药物、金属离子等[12 - 14]的分析。本研究建立了一种基于罗丹明B(RhB)与AuNPs荧光共振能量转移测定卡托普利的荧光分析新方法,用于卡托普利药片中卡托普利含量的测定,取得了满意结果。
1 实验部分
1.1 仪器与试剂
F-7000型荧光分光光度计(日本,日立公司);JEM-2100型透射电镜(日本,日立公司);TU1901 紫外-可见分光光度计(普析光学仪器有限公司);5804R型高速离心机(德国,艾本德股份公司)。
卡托普利标准溶液(4.6×10-3mol/L):准确称取卡托普利标准品(含量99.5%,上海江莱生物科技有限公司)25 mg,用超纯水溶解后定容于25 mL的容量瓶中,使用时逐级稀释。罗丹明B标准储备溶液(1.0×10-4mol/L):准确称取47.9 mg罗丹明B(分析纯,西安化学试剂厂),用超纯水溶解后,定容于100 mL 容量瓶中。氯金酸(分析纯,国药集团化学试剂有限公司);柠檬酸三钠(分析纯,西安化学试剂厂)。实验用水均为超纯水。
卡托普利药片(25 mg/tablet,上海普康药业有限公司,山西津华晖星制药有限公司)。
1.2 实验原理
以柠檬酸钠还原氯金酸得到的AuNPs被柠檬酸根保护,表面带负电荷,通过静电排斥作用使其保持稳定[16]。将AuNPs与RhB混合后,RhB分子可通过静电作用吸附在AuNPs表面,此时,由于AuNPs对RhB的荧光猝灭作用使体系呈现弱荧光;加入卡托普利后,卡托普利分子巯基上的S原子可与纳米金形成稳定的Au-S键而置换出RhB,使RhB远离AuNPs恢复其荧光,体系荧光增强。据此,通过测定加入不同浓度卡托普利后AuNPs-RhB体系荧光信号的变化,可以对卡托普利进行定量分析。卡托普利的荧光测定机理见图1。

图1 卡托普利的荧光测定机理Fig.1 Schematic diagram showing the mechanism for catopril detection
1.3 实验方法
1.3.1AuNPs的制备AuNPs按文献方法[15]进行制备。具体步骤为:将50 mL 1 mmol/L的氯金酸加入到100 mL圆底烧瓶中搅拌、加热,待溶液开始回流后快速向其中加入5 mL 38.8 mmol/L柠檬酸钠溶液,继续回流15 min后停止加热,待溶液冷却后用0.45 μm滤膜过滤,滤液置于棕色试剂瓶中,于4 ℃ 保存。
1.3.2实验方法取2.0 mL AuNPs溶液于4 mL离心管中,加入1.0 mL 1.0×10-5mol/L RhB溶液,摇匀,静态吸附1 h后,于10 000 r/min高速离心10 min,用移液枪小心移去上清液,往沉淀中加入2 mL超纯水洗涤、离心、弃去上清液,然后加入2 mL超纯水重新分散后定量转移入10 mL棕色容量瓶,适当稀释后加入2 mL柠檬酸-柠檬酸钠缓冲溶液,最后用超纯水定容,得AuNPs-RhB测定液。分别取900 μL测定液于一系列离心管中,然后依次往其中加入100 μL 超纯水、卡托普利标准品或样品溶液,放置反应1 h 后,在荧光分光光度计上测定背景(FB)和样品(FS)荧光信号,根据样品和背景的信号差△F(△F=FS-FB)测定卡托普利含量。
2 结果与讨论
2.1 AuNPs的表征及实验机理验证

图2 纳米金的吸收光谱图(内插图为TEM图)Fig.2 Absorption spectra of AuNPs(inset:TEM photogrph of AuNPs )
为了确定所制备AuNPs的尺寸和形貌,分别测了其透射电镜(TEM)和吸收光谱,结果如图2所示。图2显示合成的AuNPs在520 nm有强吸收,表明AuNPs直径为13 nm左右且无团聚[16],由TEM结果可见AuNPs粒径均一分散性良好。
为了验证我们提出的实验机理,实验分别对AuNPs、AuNPs-RhB、AuNPs-RhB-卡托普利的吸收光谱及荧光光谱进行了扫描,结果如图3所示。由图3(A)可见,加入罗丹明B及卡托普利后纳米金的吸收光谱没有发生变化,表面罗丹明B和卡托普利的加入没有引起纳米金的团聚。图3(B)为RhB及加入卡托普利前后AuNPs-RhB体系的荧光光谱,图中曲线a、b、c的峰形及最大发射波长一致,表明AuNPs、卡托普利没有与RhB作用生成新物质。但比较曲线b、c可发现加入卡托普利后体系的荧光信号明显增强,这是由于加入卡托普利后,卡托普利可通过其分子中巯基S原子与AuNPs形成稳定的Au-S共价键而置换出RhB使其远离AuNPs,减小AuNPs对其荧光的猝灭作用,从而使体系荧光强度增强。以上结果都证明了我们提出的机理,即体系荧光信号的变化是由于AuNPs对RhB荧光的猝灭作用及卡托普利对RhB的置换作用。
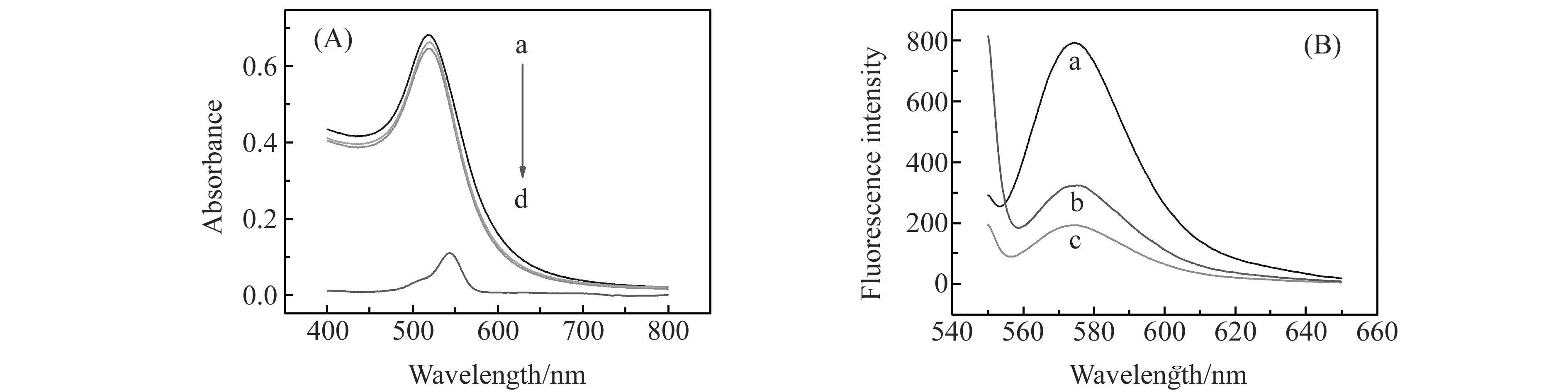
图3 吸收光谱及荧光光谱Fig.3 Absorption and fluorescence spectra(A) Absorption spectrum(a.AuNPs;b.AuNPs-RhB;c.AuNPs-RhB-catopril;d.rhodamin B);(B) Flurescence spectrum(a.RhB;b.AuNPs-RhB-catopril;c.AuNPs-RhB).
2.2 分析条件的选择

图4 pH对测定体系荧光信号变化的影响Fig.4 Effect of pH on the change of fluorescence intensity
2.2.1溶液pH的选定溶液pH是影响分析检测的重要因素,它不仅影响AuNPs和卡托普利在溶液中的存在形式,也影响Au-S键形成后体系的稳定性。实验对4.0~10.0范围内不同pH值对测定的影响进行了考察,结果如图4所示。由图4可见,不同pH值加入卡托普利后体系荧光增加值△F不同,当体系pH为6.0时△F达到最大。这可能是由于:pH值过低时,一方面RhB质子化程度高使其与AuNPs的吸附作用增强,不利于RhB从AuNPs表面脱落;另一方面,卡托普利分子中的巯基也可能质子化使Au-S键不易形成,不能有效从AuNPs表面置换RhB。而当pH过高时,卡托普利分子中的羧基可能去质子化形成-COO-,-COO-与带负电荷的AuNPs之间的静电排斥作用增强,不利于卡托普利在AuNPs表面的吸附,RhB会自发从AuNPs表面脱落,背景信号增大。
此外,实验还考察了缓冲液组成及浓度对测定影响。分别将pH为6.0的等浓度的磷酸盐、Tris-HCl、乙酸-乙酸钠和柠檬酸-柠檬酸钠缓冲溶液加入到AuNPs-RhB体系中;测定加入卡托普利后体系的荧光变化。结果发现在柠檬酸-柠檬酸钠缓冲液中体系最稳定且信噪比高;改变缓冲溶液浓度,发现柠檬酸-柠檬酸钠缓冲液浓度为5 mmol/L时测定结果最佳,故实验选择加入pH=6.0的浓度为5 mmol/L的柠檬酸-柠檬酸钠缓冲来控制体系pH。
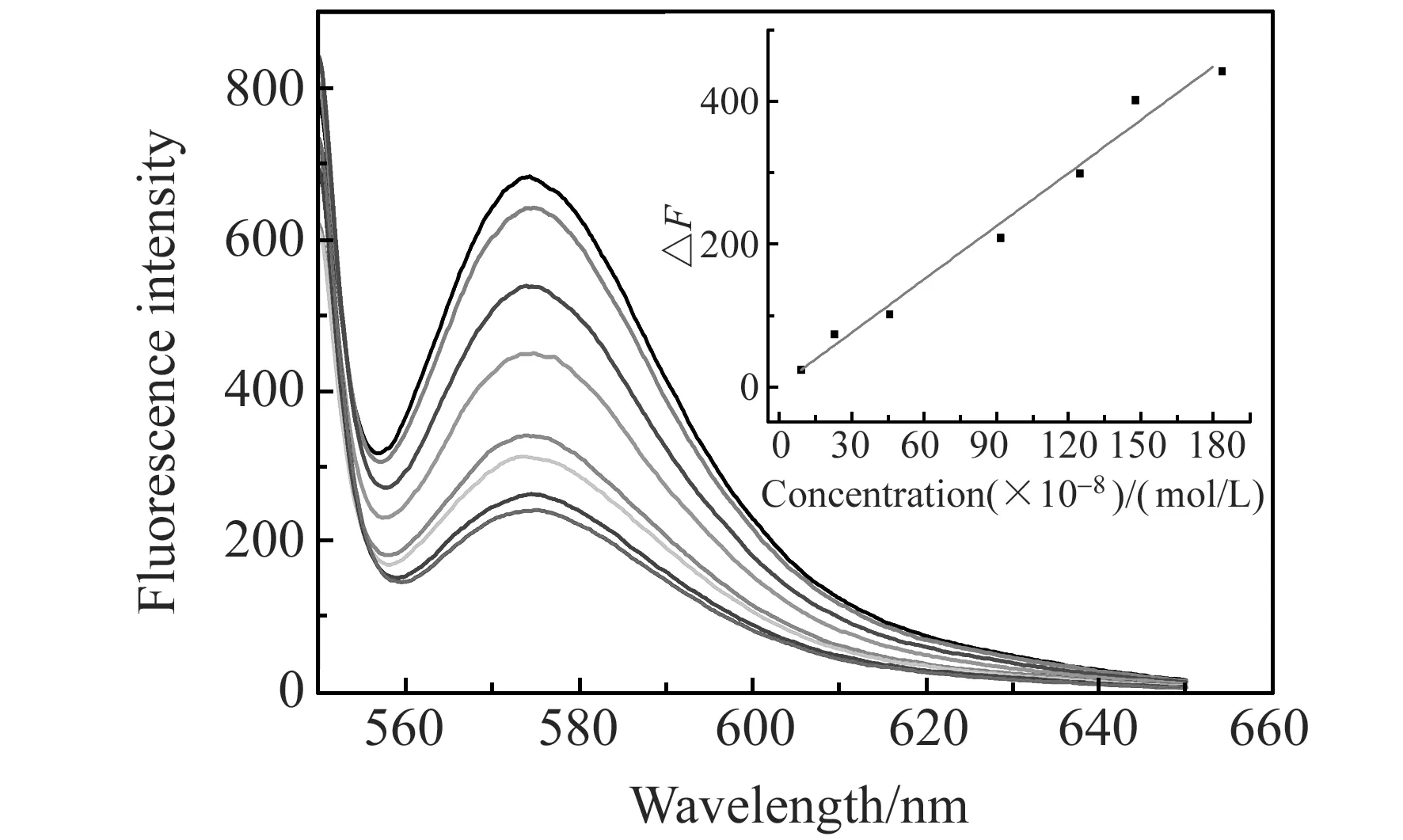
图5 不同卡托普利浓度下测定体系荧光光谱(内插图为标准曲线)Fig.5 Fluorescence spectra of AuNPs-RhB in different concentrations of catopril(inset:linearity curve) from bottom to top:0,9.2,23,46,92,125,148,184×10-8 mol/L of captopril.
2.2.2反应时间的选定取20.0 mL AuNPs-RhB溶液,加入500 μL 4.6×10-4mol/L的卡托普利标准溶液在室温下进行反应,每隔一定时间从反应体系中取出1.0 mL测定其荧光。结果表明,1 h内△F快速增大,此后△F增加缓慢,12 h后荧光信号不再增加。结合考虑测定灵敏度和实验效率选择反应时间为1 h。
2.2.3AuNPs和RhB浓度的选择AuNPs和RhB的浓度直接影响测定的灵敏度,由于游离RhB自身有很强的荧光,如果其浓度太高,会使背景信号增大;反之,若RhB浓度太低,则其在AuNPs表面吸附量不够,加入卡托普利后不能与其发生有效置换,使测定灵敏度降低。因此,只有在二者之间的平衡态时,测定才最灵敏,为此实验考察了AuNPs和RhB的浓度对测定结果的影响。实验中我们将一定量AuNPs与足够量的RhB混合作用后,通过离心洗涤除去过量的罗丹明B,然后将得到的AuNPs-RhB体系稀释一定倍数后进行测定,结果显示AuNPs与RhB(1.0×10-5mol/L)按体积比2∶1混合作用,稀释5倍后测定灵敏度最高。
2.3 标准曲线及检出限
优化实验条件下,对卡托普利进行测定,结果表明在9.2×10-8~1.8×10-6mol/L浓度范围内体系的△F与卡托普利的浓度呈良好的线性关系(图5),线性方程为:△F=2.4728c-1.0515,相关系数r=0.9929,检出限为6.9×10-8mol/L。优化的实验条件下,对2.3×10-7mol/L卡托普利标准溶液平行测定,其相对标准偏差为1.32%(n=11)。
2.4 干扰的测定
在优化的实验条件下,考察了可能存在的添加剂对卡托普利测定的影响,结果表明:控制相对误差在±5%以内时,同浓度的葡萄糖、抗坏血酸、糊精、乳糖、蔗糖、淀粉,20倍的Mg2+、Ca2+、Zn2+、Fe3+均无干扰。因此药物中常用的助剂、添加剂及一些常见金属离子对卡托普利的测定无影响。
2.5 样品测定
随机抽取10片卡托普利药片,研细混匀后准确称取适量(相当于卡托普利25 mg),加入去离子水溶解后,转入25 mL容量瓶中定容、静置,依次用滤纸和0.45 μm滤膜过滤,按实验方法测定。测定结果表明该法测得的卡托普利含量与药品的标示量基本一致。为了进一步验证方法的准确性,采用标准加入法测定了样品的回收率,测得回收率在96.5%~106.5%之间,表明该法用于卡托普利片剂中卡托普利含量的测定结果准确。
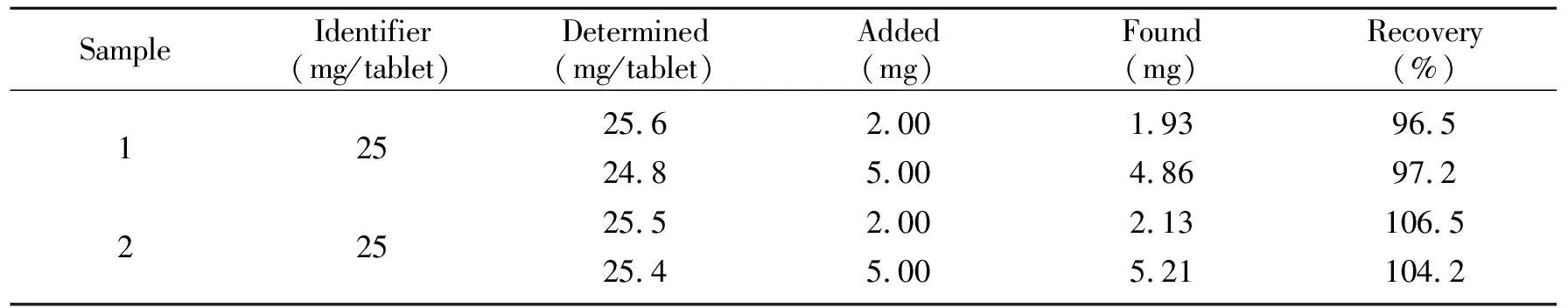
表1 样品及回收率测定(n=3)
3 结论
在罗丹明B与纳米金粒子的混合体系中,金纳米粒子可通过静电作用与罗丹明B结合,使其荧光猝灭,加入卡托普利后,金纳米粒子与卡托普利形成稳定Au-S键而置换出罗丹明B,使罗丹明B远离金纳米粒子而恢复其荧光。据此建立了罗丹明B金纳米粒子荧光共振能量转移测定卡托普利的荧光分析新方法,该方法能够快速、灵敏的用于药物中卡托普利含量的检测。
