结合QSAR、分子对接和动力学模拟剖析小分子与不同受体的结合亲和力
2016-10-16伍智蔚易忠胜冯理涛
伍智蔚, 易忠胜*, 董 露, 冯理涛
(广西高校食品安全与检测重点实验室,岩溶地区水污染控制与用水安全保障协同创新中心,桂林理工大学化学与生物工程学院,广西桂林 541004)
多溴二苯醚(Polybrominated Diphenyl Ethers,PBDEs)是一类阻燃添加剂,已广泛应用于商业和家用产品[1]。近年来,在动植物体[2 - 4]中均检出PBDEs,而通过食物链PBDEs最终会在人体内蓄积[5 - 7]。值得注意的是,PBDEs对人体造成不同的毒性作用[8,9]。此外,PBDEs经过体外代谢和藻类植物可以转化为毒性更高的羟基化物(OH-PBDEs),从而造成二次污染[10]。因此,研究PBDEs和OH-PBDEs的各项毒性指标变得越来越重要。PBDEs和OH-PBDEs的标准样品难以获取且实验成本很高,目前仅有少部分的PBDEs同系物被测定各项活性指标。定量构效关系方法(QSAR)和分子对接技术可在有限的实验数据和晶体模型基础上,利用数学建模方法建立基于PBDEs或OH-PBDEs分子结构特征的各项生物活性的预测模型,能够有效评估PBDEs或OH-PBDEs同系物的各项生物活性及机理[11]。然而,QSAR局限于大数据量目标物的构效分析,分子对接由于其分子的刚性对接模型具有不确定性[12]。
本文在前人的工作基础上,分别利用比较分子相似性指数法(CoMSIA)和分子全息定量构效关系法(HQSAR)构建模型,研究PBDEs和OH-PBDEs对芳基碳氢化合物亲和力受体(AhR)和甲状腺转运蛋白(TTR)的毒性亲和力,并结合分子对接[13]剖析其相关结合机理。此外,通过分子动力学模拟(MD)技术,探讨模型结合动态模拟过程。本文从静态和动态两个方面分别探讨AhR和TTR的亲和力,有效避免了QSAR和分子对接的缺陷,不仅建立了具有高预测能力模型还可视化地解释结合的过程。
1 原理和方法
1.1 数据来源
18个芳烃受体(AhR)的活性数据[14]和28个甲状腺转运蛋白(TTR)的活性数据均来自文献[15 - 17],活性数据采用相对亲和力(RBA)的负对数pI表示。为保证建立模型样本集结构多样性,两组数据集均按照活性值由大到小的顺序排列,并且按每隔两个数集选取一个数集作为测试集,其余数集为训练集,对模型进行内部检验。
1.2 小分子表征及预处理
整个建模过程均在Tripos公司的sybyl(版本,x-1.1)软件包中进行。首先利用Chemdraw(版本,2005)构建小分子结构,并对每个分子在Tripos力场中使用Powel能量梯度法进行能量最小化。然后以二苯醚为骨架对小分子进行叠合,使用LOO方法确立最佳主成分数并以偏最小二乘法(PLS)建立立体、疏水和静电场的CoMSIA模型,同时采用HQSAR法建立模型比较PBDEs及OH-PBDEs对不同受体的毒性亲和力贡献。
1.3 分子对接与分子动力学模拟
为研究各小分子与受体的作用模型,本文利用sybyl(版本x-1.1)中的Surflex-dock模块对小分子与受体进行对接。其中受体复合物晶体结构(ID:4M4X和4PM1)从Protein Data Bank(PDB)中获得,加氢,然后使用AMBER方法计算原子电荷。以CH4、N-H和C=O为分子探针(阀值为0.5,膨胀系数为1),确定受体空腔并进行对接。合并受体和活性最高的小分子,进行随后的动力学模拟研究,并使用do_dssp程序对动力学模拟结果进行二级结构分析。
2 结果与讨论
2.1 CoMSIA和HQSAR模型建立
分别对两组样本集进行CoMSIA模型分析,结果如表1所示。交叉验证相关系数q2均大于0.55,表明模型具有较好的预测能力;非交叉检验的PLS回归得到拟合相关系数r2均达到0.95以上,标准偏差(SEE)也均小于0.35,说明模型均具有一定的稳定性。模型中立体场(S)、疏水场(H)和电子场(E)的贡献结果见表1。在AhR受体中模型的场效应贡献主要来源于疏水场(36.9%)和电子场(54.2%),说明范德华作用对模型有较大影响,小分子的大小对于复合物的结合没有多大影响;而在TTR受体中上述三个场的贡献相对平均,分别为27.1%(立体场)、34.6%(疏水场)和38.3%(电子场)。
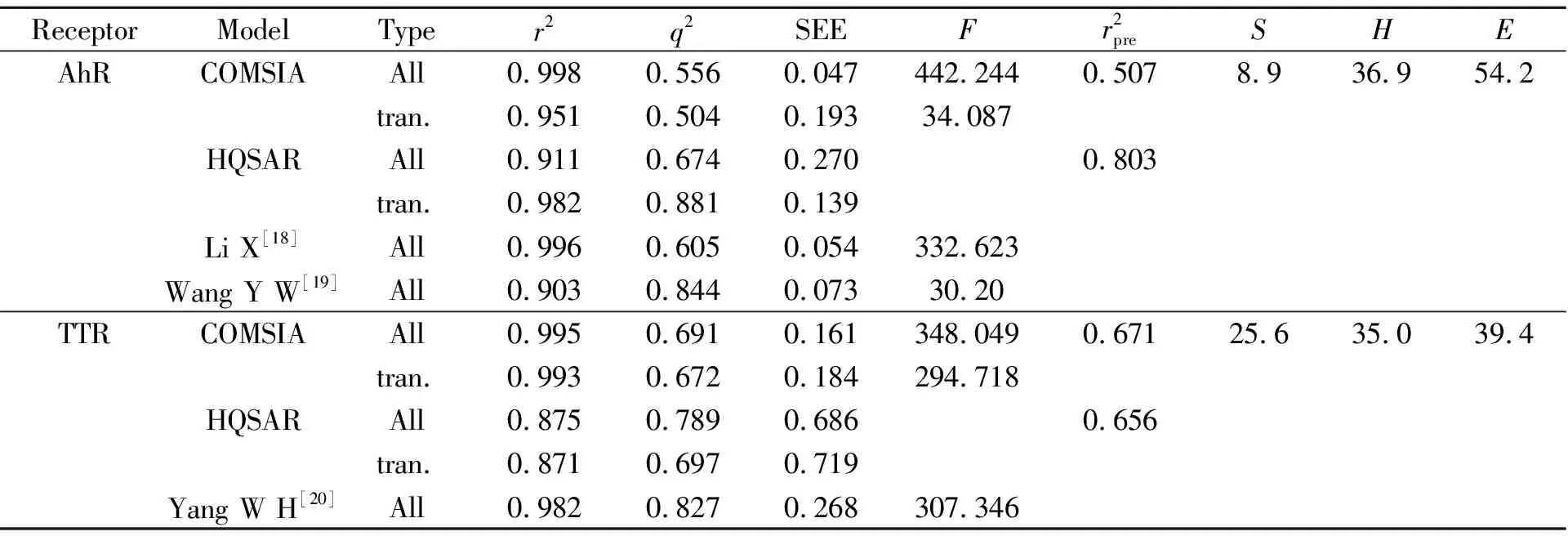
表1 模型结果和比较
同时,在分子碎片大小为4~7不变的情况下尝试不同分子碎片区分参数组合建立HQSAR模型。结果表明:AhR样本集选择碎片区分参数为原子类型(Atom, A)、化学键类型(Bonds,B)、连接性(Connections,Co)和氢原子(Hydrogenatom,H)时,可得到最佳模型:r2=0.911,q2=0.674,该模型具有较高的预测能力和稳定性。此外,TTR的碎片区分参数选择A+B+Co和氢键供/受体(Donor & Acceptor,DA)时有最佳模型(r2=0.875),模型的预测能力较AhR的模型大,q2=0.789。由此可推测小分子与TTR结合时氢键对复合物结合模式的影响较大。
2.2 模型预测能力和稳健度检验
2.3 模型分析
以活性最高化合物01(AhR:BDE154,TTR:BDE19)为例对模型进行等高线图分析,如图1所示。a(d)、 b(e)和c(f)分别为AhR(TTR)受体的CoMSIA模型的立体场图、静电场图和疏水场图。从立体场图中可以看出C6′(a)和C2(d)位置存在大块的黄色区域,说明此位置引入大体积的基团可以提高化合物的活性。而C3′(a)位置绿色区域引入小体积基团有利于提高化合物活性,如AhR模型中BDE154(pI=4.64)在C6′位置存在溴,C3′无取代基,其活性比溴取代基位置相反的BDE119(pI=2.98)活性大,因此BDE119对AhR的亲和力更强。在图1b(e)中,蓝色区域表示引入带正电荷基团有利于提高化合物的活性,红色区域表示引入带负电基团有利于提高化合物的活性。图1b中C5′和C3附近有蓝色区域,同样图1e中C4位置也有蓝色区域,这都说明这些区域引入带正电基团有利于提高活性。同时,在疏水场中,红色区域引入疏水性基团有利于提高化合物活性,也就是说亲水性基团将会致使化合物活性降低。例如,TTR受体疏水场(图1f)中C4′位置周围有黄色区域,当该位置被具有强亲水基团取代,化合物活性会降低。因此BDE49(pI=2.55)活性比4′-OH-BDE49(pI=-0.48)活性大,说明带羟基的化合物对TTR受体的亲和力较大。
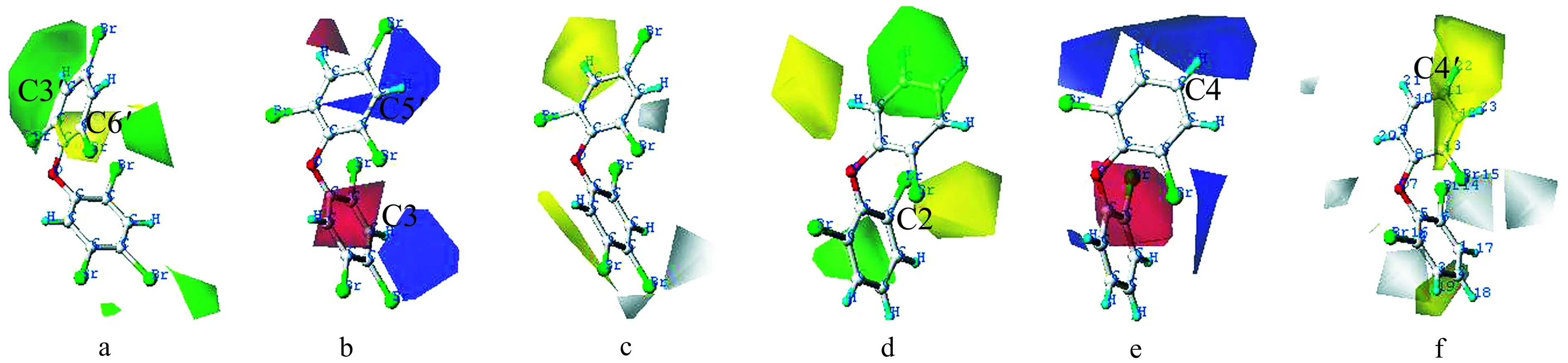
图1 模型的等高线图Fig.1 Contour maps of CoMSIA model
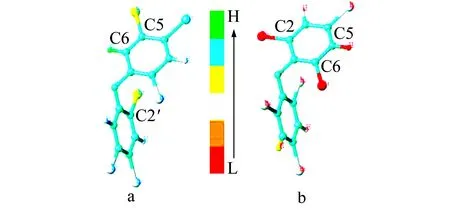
图2 HQSAR模型的原子对化合物活性贡献图Fig.2 Contribution to compound activities of different atoms in HQSAR model
同时,图2给出了HQSAR预测模型中原子对化合物活性的贡献情况。图2中的原子颜色表明该原子对预测模型的贡献,由低到高依次为:红橙白黄蓝绿。处于光谱中红色系的颜色反映的是不利的贡献,从橙色到红色这种不利的影响依次增大,处于该色系的基团对受体的亲和力贡献较小,如图2b中C2和C6上的溴取代基以及C5位上的氢原子;反之处于光谱蓝色系的颜色反映为有利的贡献,从黄色到绿色表示对活性的贡献依次增大,促使具备该色系基团的小分子与受体间的亲和力增大,如图2a中C5和C2′上的溴原子以及C6上的氢原子。
2.4 分子对接结果
分子对接后可以产生多个结合构象,我们选择结合能量最低构象作为目标构象。分别以分子活性最高分子01与受体(AhR和TTR)对接结果为例进行详细讨论。图3给出了两个受体与小分子6 Å范围内的结合情况。在AhR受体中,BDE154完全进入到疏水空腔中,被受体残基包围,形成稳定的结合构象。如小图a所示,小分子BDE154与附近残基Leu110,Met109和Tyr135形成疏水作用。而在TTR受体中,小分子BDE019只在受体的表面结合。但是BDE019周围的残基与其形成π-π堆积作用的残基屋顶结构,见小图b,使得受体TTR与小分子的结合变得更稳定。同时,从a中可以看出BDE019的两个苯环分别被残基Leu17、Lys15、Leu110、Ala109、Ala108、Thr119和Val121所包围,主要为疏水作用。说明在此位置引入疏水作用强的基团有利于提高化合物的活性。

图3 分子对接结果Fig.3 The results of molecular docking
2.5 动力学模拟结果
使用分子动力学模拟(MD)的方法,选择相应活性最高的小分子(AhR:BDE154,TTR:BDE19)作为配体,对AhR和TTR的结合模型进行10 ns模拟。采用do_dssp程序分别对AhR和TTR的分子动力学模拟结果进行估算,结果如表2所示。当小分子与AhR或者TTR结合时,蛋白受体的二级结构都发生了相互间的转化,而这种转化则有可能是促使蛋白与小分子结合得更稳定的一种可视化的表现。比如当小分子BDE154进入到AhR中时,β-折叠(B-Sheet)、α-螺旋(A-Helix和3-Helix)的含量百分比减少,弯曲(Coil)和转角(Turn)则增加。而在TTR与BDE19的结合模型中,这产生了相反的结果。当小分子BDE154进入到AhR中时,β-折叠(B-Sheet)含量百分比增加,而弯曲(Coil)的含量百分比减少。这说明不同受体与小分子结合时,会以不同的结构转化方式进行结合,而这种转化方式是随着受体和配体的不同而改变的。
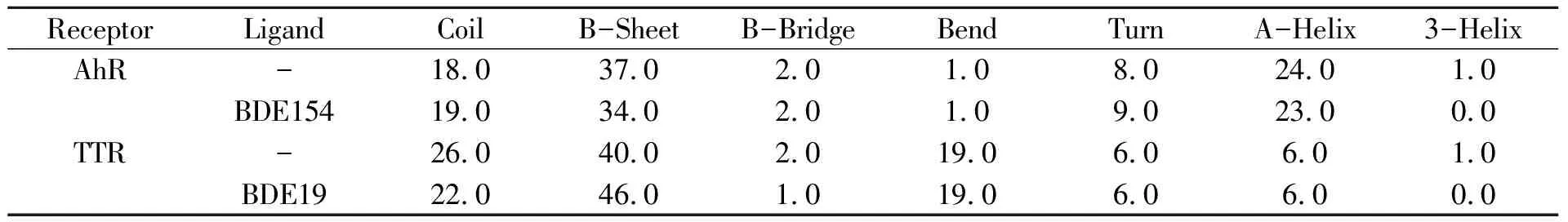
表2 受体蛋白二级结构平均估算值(%)
3 结论
QSAR、分子对接和分子动力学模拟三种方法用于研究多溴二苯醚及其羟基化物与不同受体(AhR和TTR)的亲和力构效关系,分别得到了具有较高q2和r2的模型。从配体的原子附近三维等值线图;不同原子对活性的贡献图;疏水空腔对接图三个不同角度分析小分子与受体的结合机制,并且结果较为一致,并且在动力学模拟结果的二级结构中分析发现随着小分子的加入,受体的二级结构发生转变,而这种转变使得结合模型更加稳定。三种方法相互补充,为指导研究小分子与受体结合研究提供有效的分析手段和预测模型。
