HPLC法同时测定磷酸哌喹有关物质与含量
2016-10-12黄军
黄 军
(桂林南药股份有限公司,桂林 541004)
HPLC法同时测定磷酸哌喹有关物质与含量
黄 军
(桂林南药股份有限公司,桂林 541004)
目的:建立同时测定磷酸哌喹有关物质与含量的HPLC方法。方法:采用反相高效液相色谱法,测定磷酸哌喹有关物质与含量,并分别计算其杂质A~I的相对保留时间及校正因子。色谱条件:采用Welch Ultimate XBC18(100mm×4.6mm,3μm)柱,以0.1%三氟乙酸水溶液为流动相A,以乙腈为流动相B,线性梯度洗脱。流速1.5ml·min-1,柱温35℃,检测波长240nm。结果:磷酸哌喹与其各杂质峰分离度良好;磷酸哌喹与杂质A~I在各自线性范围内线性关系良好(r=0.9999); 磷酸哌喹,杂质A~I的检测限分别为3.4 ng·ml-1、1.0 ng·ml-1、4.9 ng·ml-1、3.1 ng·ml-1、1.4 ng·ml-1、3.8 ng·ml-1、1.7 ng·ml-1、2.8 ng·ml-1、1.9 ng·ml-1、2.0 ng·ml-1;杂质A~I加样回收率在95.5%~99.4%之间,RSD(n=9)分别为2.8%、2.7%、1.9%、3.5%、1.8%、1.8%、1.8%、2.5%、1.1%;3批样品含量测定结果分别为99.9%,99.6%和99.8%。供试品溶液在24小时内稳定。结论:本方法灵敏度好,操作简便,结果准确,可作为磷酸哌喹的质量控制。
磷酸哌喹;HPLC;有关物质;含量
磷酸哌喹用于疟疾的治疗,也可作症状抑制性预防用,尤其是耐氯喹虫株恶性疟的治疗与预防[1]。因磷酸哌喹单方制剂易产生抗药性,磷酸哌喹通常与双氢青蒿素、甲氧苄啶等组成复方制剂使用,如磷酸哌喹与双氢青蒿素合用,具有显著的协同增效作用,可延缓疟原虫抗药性的产生[2-6]。
目前关于磷酸哌喹含量测定的报道较多见[7-12],但磷酸哌喹有关物质的检测方法则鲜有报道,且现有的报道中未见对杂质进行定性和定量[13-15]。现行的《中国药典》2015年版二部中磷酸哌喹有关物质采用的是高效液相色谱法,但只对3个已知杂质进行控制,含量测定则采用滴定法[16]。随着人们对药品安全问题越来越关注,今后对药物杂质的控制要求也将越来越严格。笔者经过摸索,建立了磷酸哌喹有关物质与含量测定的高效液相色谱方法。该法采用加校正因子的主成分自身对照法,可对磷酸哌喹原料中可能出现的9个已知杂质进行较准确的控制,同时可以用于测定磷酸哌喹的含量。
1 仪器、试剂与方法
1.1仪器
高效液相色谱仪:日本岛津LC-20A,紫外检测器;安捷伦1260,DAD检测器。紫外可见分光光度计:北京普析,TU-1901。电子天平:瑞士梅特勒,XS105。Mill-Q超纯水器:美国密理博公司。
1.2药品与试剂
磷酸哌喹对照品为中国药品生物制品检定所对照品(批号100751-200501,含量91.2%),磷酸哌喹的杂质A(7-氯-4-(1-哌嗪基)喹啉,7-Chloro-4(1-piperazinyl)quinoline,含量98.9%)、杂质B(7-氯-4-(4-(3-(哌嗪-1-基)丙基)哌嗪-1-基)喹啉,7-Chloro-4-(4-(3-(piperazin-1-yl)propyl)piperazin-1-yl)quioline,含量98.6%)、杂质C(1-[4-(7-氯喹啉-4-基)哌嗪-1-基]-3-[(4-(5-氯喹啉-4-基)哌嗪-1-基]丙烷,1-[4-(7'-chloro-quinoline-4')piperazine]-3-[4-(5'-chloro-quinoline-4')piperazine-1]-propane,含量98.2%)、杂质D(7-氯-4-羟基喹啉,7-Chloro-4-hydroxy-quinoline,含量98.8%)、杂质E(1,4-二(3-(4-(7-氯喹啉-4-基)哌嗪-1-基)丙基)哌嗪,1,4-bis[3-(4-(7'-chloro-quinoline-4')pierazine-1)propyl]pipeazine,含量98.1%)、杂质F(7-氯-4-(4-(3-((7-氯喹啉-4-基)氧代)丙基)哌嗪-1-基)喹啉,7-chloro-4-(4-3-((7'-chloro-quinoline-4')oxo)propyl)pipe razine-1)quinoline,含量98.7%)、杂质G(N1-(7-氯喹啉-4-基)-N2-(3-(4-(7-氯喹啉-4-基)哌嗪-1-基)丙基)乙烷-1,2-二胺,N1-(7'-chloro-quinoline-4')-N2-(3-(4-(7'-chloro-quinoline-4')piperazine-1)propyl)ethane-1,2-diamine,含量99.0%)、杂质H(1,4-二(7-氯喹啉-4-基)哌嗪,1,4-Di(7-chloroquinoline-4-yl)piperazine,含量99.8%)、杂质I(4,7二氯喹啉,4,7-Di-quinoline,含量99.2%)对照品均由某公司提供,磷酸哌喹原料药(桂林南药股份有限公司,批号150401、150402、150403,水分分别为7.4%、7.2%、7.1%)。乙腈(色谱纯,批号4292053,Dikma Pure),三氟乙酸(色谱纯,批号14020696,TEDIA),水为Milli-Q制备的纯化水。
1.3色谱条件
色谱柱为Welch Ultimate XB-C18(100mm×4.6mm,3μm);检测波长:240nm;流速:1.5ml·min-1;柱温:35℃;进样量:20μl;流动相:流动相A为0.1%三氟乙酸水溶液,流动相B为乙腈,按表1进行梯度洗脱。

表1 梯度洗脱程序Tab 1 HPLC gradient program
1.4溶液的配制
避光操作。溶剂:水-乙腈-三氟乙酸(850:150:1)。
有关物质测定溶液的配制:取磷酸哌喹约25mg,精密称定,置50ml容量瓶中,加溶剂溶解并稀释至刻度,得到浓度约为0.5mg·ml-1的溶液,作为供试品溶液。精密量取1ml 置100ml容量瓶中,用溶剂稀释至刻度,摇匀,作为对照溶液。
含量测定溶液的配制:取磷酸哌喹约32mg,精密称定,置50ml容量瓶中,加溶剂溶解并稀释至刻度,摇匀,精密量取1ml,置20ml容量瓶中,加溶剂稀释至刻度,摇匀,得到浓度约为32μg·ml-1的溶液。
各杂质对照品溶液及系统适用性溶液的配制:取杂质A~I对照品各约10mg,精密称定,分别置100ml容量瓶中,加溶剂适量溶解并稀释至刻度,作为各杂质对照品溶液。取磷酸哌喹对照品约25mg,精密称定,置50ml容量瓶中,精密量取各杂质对照品溶液2.5ml加入,加溶剂溶解并稀释至刻度,摇匀,作为系统适用性溶液。
1.5方法学考察实验(避光操作)
1.5.1标准曲线 取磷酸哌喹对照品约10mg,精密称定,配制成浓度约为0.1mg·ml-1的溶液。取上述溶液及“1.4”项下各杂质对照品溶液,倍比稀释得到6个不同浓度的标准液,在本色谱条件下进样,结果与定量限一起计算。以溶液浓度C(μg·ml-1)为横坐标,峰面积A为纵坐标,绘制标准曲线。
1.5.2重复性试验 取150401批磷酸哌喹供试品12份,取其中6份按“1.4”项下方法配制成有关物质测定溶液,另6份配制成含量测定溶液。取磷酸哌喹对照品约32mg,配制成含量测定浓度的溶液,作为含量测定对照品溶液。取上述溶液进样,计算试验结果。
1.5.3回收率试验 取杂质A~I对照品各3份,约为10mg,精密称定,分别置100ml容量瓶中,加溶剂适量溶解并稀释至刻度,作为各杂质对照品储备液。精密量取各个杂质的1份储备液50μl加入同一装有约25mg磷酸哌喹(批号:150401)的50ml容量瓶中,加溶剂超声溶解并稀释至刻度,摇匀,得到低浓度杂质的测试溶液,再分别精密量取各个杂质剩下的2份储备液各250μl、500μl,按上述低浓度杂质测试溶液的配制方法,分别配制成中、高浓度杂质测试溶液。每种浓度测试溶液平行配制3份,共9份。另取各杂质约10mg,精密称定,按中浓度杂质测试溶液配制方法配制成溶液,作为对照溶液。取上述溶液进样,计算回收率结果,磷酸哌喹已知杂质量按重复性试验项下的平均杂质量计。
1.5.4破坏实验 取150401批磷酸哌喹3份,每份约25mg,精密称定,分别置50 ml量瓶中。第1份加1 mol·L-1盐酸2ml,超声30秒钟摇匀,于室温避光放置24小时后,加2ml的1mol·L-1氢氧化钠溶液中和,用溶剂稀释至刻度,摇匀,作为供试品酸破坏试验溶液,同时做空白试验;第2份加1 mol·L-1氢氧化钠溶液2ml,超声30秒钟摇匀,于室温避光放置24小时后,加2ml的1 mol·L-1盐酸中和,用溶剂溶解并稀释至刻度,摇匀,作为供试品碱破坏试验溶液,同时做空白试验;第3份加3%过氧化氢溶液2ml,超声30秒钟摇匀,于室温避光放置15小时后,强烈振摇15min,用溶剂溶解并稀释至刻度,摇匀,作为供试品氧化破坏试验溶液,同时做空白试验。取上述溶液分别进样,记录色谱图。
2 结果与讨论
2.1波长的选择与校正因子
2.1.1波长的选择
取杂质A~I及磷酸哌喹对照品各适量,分别用流动相溶解并稀释成浓度约为5μg·ml-1的溶液。按紫外-可见分光光度法,分别在200nm~400nm波长段内扫描,磷酸哌喹在226nm、239nm、347nm波长处有最大吸收,其各杂质均在240nm波长附近有最大吸收。综合上述紫外波长扫描结果,本法选择240nm作为检测波长。在选择的波长上,与现行中国药典及多数文献的波长有所不同。现行中国药典有关物质检测分别采用的是317nm与349nm两个波长,含量测定采用349nm波长。
2.1.2校正因子
因磷酸哌喹与其杂质的结构有差异,其各自的紫外波长吸收情况也有所不同,如仅用主成分自身对照法很难准确测定各杂质含量,故有必要根据情况引入校正因子。另外,磷酸哌喹的杂质个数较多,如使用外标法测定杂质含量,需要用到多个杂质对照品,检测成本较高。本文采用加校正因子的主成分自身对照法,可降低检测成本,亦可较准确的测定各杂质含量。
2.2流动相的选择
在现行中国药典中,流动相使用三氯乙酸作为缓冲液,其他文献则多用磷酸盐作为缓冲液。笔者在通过试验比较,发现使用三氟乙酸代替三氯乙酸及其他缓冲盐,磷酸哌喹杂质的分离效果更好,并且三氟乙酸能在一定程度上改善色谱峰的峰型,防止峰展宽或者拖尾,故选择三氟乙酸作为缓冲液。
2.3溶液配制的操作
因磷酸哌喹遇光易变色,为避免配制的溶液遇光产生杂质,其有关物质溶液在配制时采取避光操作,这点现行中国药典也有说明。本实验的操作,在无日光灯直射的条件下进行,并使用棕色容量瓶代替透明容量瓶。
2.4杂质结构式
对磷酸哌喹原料进行杂质质量控制时,其合成起始物料及合成过程中产生的杂质均根据需要作为杂质一起控制。本法控制的9个杂质结构式见图1。
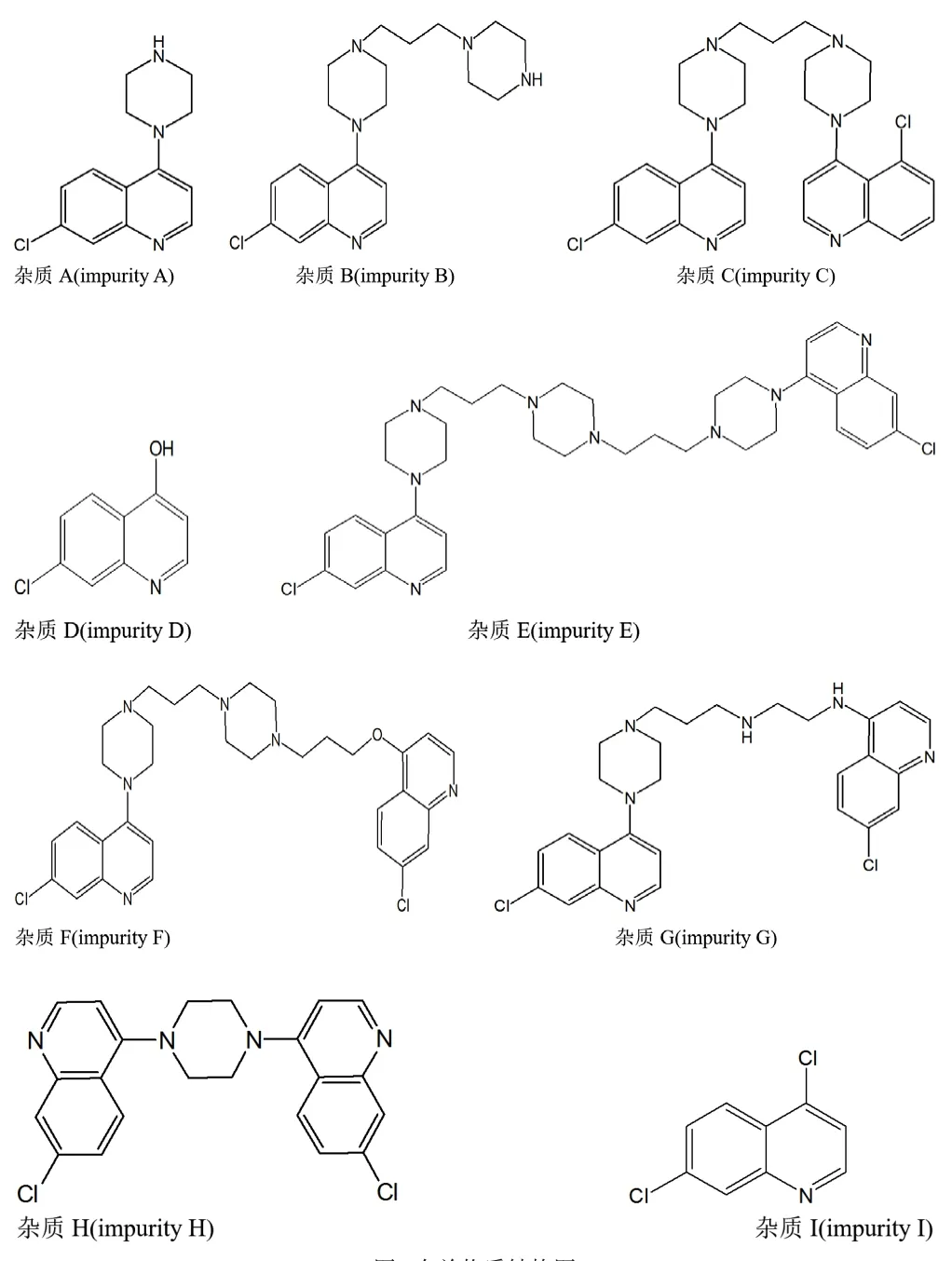
图1 有关物质结构图Fig 1 The structures of impurities
2.5方法学考察结果与样品测定
2.5.1专属性
2.5.1.1空白溶剂干扰和杂质分离度试验
取空白溶剂、各杂质对照品溶液及系统适用性溶液分别进样,记录色谱图,见图2。试验结果,空白溶剂对磷酸哌喹及其杂质检测无干扰。杂质A~I的相对保留时间分别为0.21、0.26、0.95、0.92、1.05、1.18、1.16、1.46、1.88。各相邻成分色谱峰之间分离度均大于1.5,磷酸哌喹峰理论塔板数为22273。
2.5.1.2破坏实验
将供试品破坏实验色谱图与重复性试验项下色谱图相比较。破坏实验结果,空白试验溶剂峰对杂质峰的检测无影响;在本实验酸、碱破坏条件下,色谱图中磷酸哌喹主峰面积未发生明显变化,杂质峰未见增大,且未见有新的杂质峰生成,说明磷酸哌喹对酸、碱较为稳定;在氧化破坏条件下,磷酸哌喹主峰面积减少,破坏产生的主要产物为相对保留时间约为1.11的未知杂质,以及少量的杂质B和杂质D(见图3)。色谱图中各成分色谱峰的分离度均大于1.5,主要降解峰、磷酸哌喹峰纯度因子均在990阈值限值内,为纯峰。以上专属性试验结果表明,本法专属性良好。
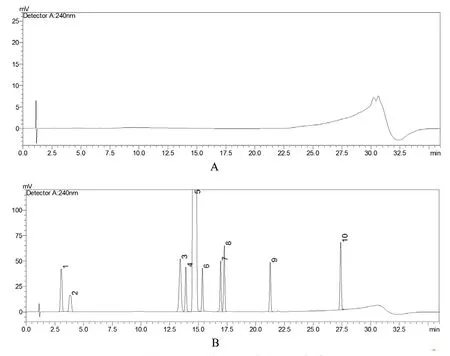
图2 溶剂(A)、系统适用性溶液(B)色谱图Fig 2 Chromatograms of solvent(A)and suitability(B)

图3 破坏试验(氧化)色谱图Fig 3 Chromatogram for destruction test(oxidation)of piperaquine phosphate
2.5.2检测限、定量限
取磷酸哌喹对照品溶液及各杂质对照品溶液,逐步稀释,在信噪比S/N约为10时,测得磷酸哌喹、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G、杂质H、杂质I的定量限分别为10.4ng·ml-1、3.0ng·ml-1、14.6 ng·ml-1、9.2 ng·ml-1、4.3 ng·ml-1、11.4 ng·ml-1、5.0 ng·ml-1、8.4 ng·ml-1、5.6 ng·ml-1、5.9 ng·ml-1;在信噪比S/N约为3时,测得磷酸哌喹、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G、杂质H、杂质I的检测限分别为3.4 ng·ml-1、1.0 ng·ml-1、4.9 ng·ml-1、3.1 ng·ml-1、1.4 ng·ml-1、3.8 ng·ml-1、1.7 ng·ml-1、2.8 ng·ml-1、1.9 ng·ml-1、2.0 ng·ml-1。
2.5.3标准曲线及校正因子
磷酸哌喹斜率与各个杂质的斜率的比值即为各个杂质的校正因子,由此计算得到各个杂质的校正因子。绘制的标准曲线结果及各杂质校正因子见表2。
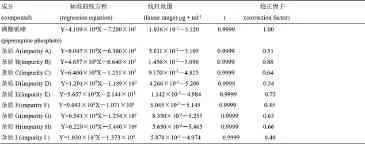
表2 磷酸哌喹及9个杂质的标准曲线方程、线性范围、相关系数、校正因子Tab2 Regression equations,linear ranges,correlation coefficients,correction factors of piperaquine phosphate and nine impurities
结果表明,在各自的线性范围内,磷酸哌喹及9个杂质的峰面积与其浓度均呈良好的线性关系。
2.5.4重复性试验
试验结果,有关物质6份供试品的总杂质分别为0.15%、0.14%、0.15%、0.14%、0.15%、0.15%,RSD为3.5%,小于10%;含量测定6份供试品磷酸哌喹的含量分别为99.8%、99.6%、99.8%、100.4%、99.9%、100.0%,RSD为0.3%。结果表明有关物质及含量测定重复性良好。
2.5.5回收率试验
结果计算得到杂质A~I的平均加样回收率(n=9)分别为96.6%、95.5%、99.1%、97.4%、98.4%、98.4%、98.5%、98.6%、99.4%,RSD分别为2.8%、2.7%、1.9%、3.5%、1.8%、1.8%、1.8%、2.5%、1.1%。说明本法准确度良好。
2.5.6溶液稳定性试验
取重复性试验项下的一份含量测定供试品溶液,于室温放置4、8、12、18、24h后,按本方法色谱条件进样。结果与该溶液临用新配的进样结果一起计算,计算得磷酸哌喹峰面积的RSD为0.5%。表明磷酸哌喹供试品溶液在24小时内稳定。
取新配制的系统适用性溶液,按本方法色谱条件进样,再于室温分别放置4、8、12、18、24h后进样。结果计算得磷酸哌喹杂质A~I峰面积的RSD分别为0.6%、0.3%、0.5%、0.1%、0.2%、0.4%、0.3%、0.6%、0.5%。表明磷酸哌喹杂质溶液在24h内稳定。
2.5.7样品测定
2.5.7.1样品含量测定
取150401批、150402批、150403批供试品及磷酸哌喹对照品,按“1.4”项下方法分别配制含量测定溶液和相同浓度的含量对照品溶液,按本法色谱条件进样,记录色谱图。按外标法以峰面积计算含量。结果见表3。

表3 样品含量测定结果(n=3)Tab 3 The sample determination results
2.5.7.2有关物质测定
取150401批、150402批、150403批供试品,按“1.4”项下方法分别配制有关物质测定溶液,按本法色谱条件进样,记录色谱图。结果按加校正因子的主成分自身对照法计算各杂质的含量。结果见表4。

表4 磷酸哌喹有关物质测定结果(%)Tab 4 Determination of related substances of piperaquine phosphate
3 结论
本文建立的高效液相色谱方法,作为有关物质测定可同时控制9个磷酸哌喹已知杂质,亦可用于磷酸哌喹的含量测定。该法灵敏度高,结果准确,操作简单,可作为磷酸哌喹原料药质量控制的参考。
[1] Chinese Pharmacopoeia committee(中华人民共和国药典委员会). The Notice of Drugs in Clinical Use of ChP(中华人民共和国药典临床用药须知)[M].Beijing(北京): China Medical Science Press(中国医药科技出版社),2010:858~858.
[2] C Lon,JE Manning,P Vanachayangkul,et al. Efficacy and effectiveness of dihydroartemisinin-piperaquine versus artesunatemefloquine in falciparum malaria: an open-label randomised comparison[J]. Plos One,2014,9(3): e93138-e93138.
[3] MENAN H,FAYE O,SAME-EKOBO A,et al. Comparative Study of the Efficacy and Tolerability of Dihydroartemisininpiperaquine-trimethoprim Versus Artemther-lumefantrine in the Treatmeat of Uncomplicated Plasmodium Falciparum Malaria in Cameroon,Ivory Coast and Senegal[J]. Malaria Joural,2011,10(1): 1-8.
[4] SONG Jianpin,SOCHEAT DUONG,TAN Bo,et al. Randomized Trials of Artemisinin- piperaquine,Dihydroartemisinin-piperaquine Phosphate and Artemether-lumefantrine for the Treatment of Multidrug Resistant Falciparum Malaria in Cambodia-Thailand Border Area[J]. Malaria Joural,2011,10(10): 231-236.
[5] KZ Aye,L Htet,M Barends,N Lindegardh. Efficacy and effectiveness of dihydroartemisinin-piperaquine versus artesunatemefloquine in falciparum malaria: an open-label randomised comparison[J]. Lancet,2006,367(9528): 2075-2085.
[6] TT Hien,C Dolecek,PP Mai,et al. Dihydroartemisininpiperaquine against multidrug-resistant Plasmodium falciparum malaria in Vietnam: randomised clinical trial[J]. Lancet,2004,363(9402): 18-22.
[7] 郭毅新,张元杰,丁涛. HPLC法测定双氢青蒿素哌喹片中的磷酸哌喹含量[J]. 药学研究,2014,33(6):332~333.
[8] 朱凯,范琦,张小松. HPLC测定双氢青蒿素哌喹片中磷酸哌喹的含量[J]. 中国药学杂志,2008,43(9):713~715.
[9] 李美琴,范琦,张小松. HPLC同时测定复方双氢青蒿素片中磷酸哌喹与甲氧苄啶的含量 [J]. 华西药学杂志,2004,19(3):221~223.
[10] 卢元媛,王璐,胡英杰,等. 高效液相法测定复方磷酸哌喹片中磷酸哌喹的含量[J]. 中国药学杂志,2010,45(7):551~553.
[11] 刘昌辉,黄天来,洪罄,等. 高效液相色谱法测定人血浆中磷酸哌喹[J]. 药物分析杂志,2007,27(1):63~65.
[12] 王文清,汪秋兰,张卫东,等. 高效液相色谱法测定复方磷酸哌喹片中磷酸哌喹和磺胺多辛的含量[J]. 中国医院药学杂志,2012,32(2):123~126.
[13] 张小松,江生,毛庆. 磷酸哌喹有关物质的检测研究[J].中国药事,2009,23(7):690~691.
[14] 熊苗苗,汪秋兰,施春阳,等. 高效液相色谱法测定复方磷酸哌喹片的有关物质[J]. 医药导报,2013,32(1):75~77.
[15] 周琳. HPLC测定双氢青蒿素哌喹片中的磷酸哌喹及有关物质[J]. 华西药学杂志,2015,30(5):610~612.
[16] ChP 2015.Vol Ⅱ(中国药典2015年版,二部)[S].2015:157 4~1575.
HPLC Determination of Piperaquine Phosphate Content and Its Related Substances
Huang Jun
(Guilin Pharmaceutical Co.,Ltd,Guilin,Guangxi 541004,China)
Objective: To establish an HPLC method to determine piperaquine phosphate content and its related substances. Methods: Piperaquine phosphate content and its related substances were determined by RP-HPLC. The impurity A - I in piperaquine phosphate,the relative retention times and the correction factors were determined respectively by HPLC. The chromatographic condition was as follows. The Welch Ultimate XB-C18 column(100mm×4.6mm,3μm)was adopted,the mobile phase was 0.1% Trifluoroacetic Acid in water(A)- acetonitrile(B)with gradient elution at a flow rate of 1.5 ml·min-1,the column temperature was 35℃,and the detection wavelength was 240nm. Results: Piperaquine phosphate and the impurities were separated well. The calibration curves of nine impurities were linear in each concentration range(r=0.9999). The detection limits of piperaquine phosphate and impurity A - I were 3.4 ng·ml-1,1.0 ng·ml-1,4.9 ng·ml-1,3.1 ng·ml-1,1.4 ng·ml-1,3.8 ng·ml-1,1.7 ng·ml-1,2.8 ng·ml-1,1.9 ng·ml-1,2.0 ng·ml-1,respectively. All the average recoveries of the impurity A -I were 95.5%- 99.4%,and RSDs(n=9)were 2.8%,2.7%,1.9%,3.5%,1.8%,1.8%,1.8%,2.5%,1.1%. The content determination results for the 3 batches of samples were 99.9%,99.6% and 99.8%,respectively. The solution was stable for 24h. Conclusion: The method is sensitive,easy,accurate and suitable for quality control of piperaquine phosphate.
Piperaquine phosphate; high performance liquid chromatography(HPLC); related substances; content
Q65[Document Code] A
10.11967/2016140410
Q65
ADOI: 10.11967/2016140410
黄军(1978-),男,工程师。
通讯地址:桂林南药股份有限公司技术开发部,邮编541004,Tel:(0773)8990395; E - mail: hjjj2001@163.com。
