青霉菌侵染前后哈密瓜转录组的构建及分析
2016-10-11单春会唐凤仙姜富耀
单春会, 陈 卫, 唐凤仙, 姜富耀
(1.江南大学 食品学院,江苏 无锡214122;2.石河子大学 食品学院,新疆 石河子832003)
青霉菌侵染前后哈密瓜转录组的构建及分析
单春会1,2, 陈 卫*1, 唐凤仙2, 姜富耀2
(1.江南大学 食品学院,江苏 无锡214122;2.石河子大学 食品学院,新疆 石河子832003)
丰富的营养物质容易被青霉菌侵染,从而导致哈密瓜的腐坏变质。利用Illumina测序技术考察青霉病原菌侵染前后哈密瓜基因转录组水平的差异变化。利用Illumina平台测序并分析了哈密瓜的转录组信息。过滤后的Clean Reads包含4 947 555 420 nt的碱基数,对Clean Reads进行组装,获得了50 502条Unigene。在NR数据库中,以E value值10-5为截点进行BLAST比对,共有71.28%的Unigenes比对到该数据库中。但还有10 526条Unigene没有比对到NR、NT 和Swiss-Prot数据库,可能是哈密瓜果实区别于其它物种而特有的基因,这需要进一步验证。将本次转录组测序得到的39 976个Unigene比对到KEGG数据库进行Pathway注释,共映射到不同的代谢通路128个。
哈密瓜;转录组;测序;组装结果分析
哈密瓜属于葫芦科甜瓜类植物[1],通常指的是新疆的厚皮甜瓜(Cucumis melo Lvar),是一种新疆特色瓜果。哈密瓜形态各异,风味独特,不但好吃,而且营养丰富。据张辉[2]、赵劼[3]的研究报道,哈密瓜在整个生长发育过程中均可受到病原菌的潜伏侵染,其中网纹期为病原菌侵染的较重时期。筛选并分离新疆哈密瓜采后主要致腐病原菌,并对冷藏中的果实进行潜伏侵染的真菌分离鉴定,可以得出青霉属菌等是引起哈密瓜在低温贮藏和冷藏运输中腐烂的优势病原菌[4-5]。由于哈密瓜在贮运过程中极易受到青霉病原菌的侵染,采后哈密瓜应对青霉菌侵染的反应机制和调控是提高其贮藏品质和延长贮藏时间的关键。目前转录组学己经被广泛应用于植物与微生物相互作用研究[6]。转录组是细胞或组织在特定时间或状态下所能转录出来的所有RNA的总和,是研究细胞表型和功能的一个重要手段。随着并行测序技术的发展,测序成本降低,大规模转录组测序也成为了转录 组研究的重 要方法,包括SAGE(serial analysis of gene expression),CAGE (cap analysis of gene expression),MPSS(massively parallel signature sequencing) 和 RNA-Seq(RNA sequencing)等[7-9]。近年来,转录组学在揭示植物细胞生理活动规律的研究中应用日益广泛,也分别在棉花[10]、小麦[11]、番茄[12]、大豆[13]的科研中取得了不错的应用效果。
本实验拟采用转录组学及现代分子生物信息学等技术手段,考察青霉病原菌侵染前后哈密瓜基因转录组水平的差异变化,研究采后哈密瓜对青霉病原菌侵染的应答反应。为开展相关抗病性基因的表达调控奠定基础,进而为哈密瓜采后病害的无污染防治技术和绿色保鲜技术提供理论依据。
1 材料与方法
1.1 原料
哈密瓜,新疆哈密淖毛湖农场采摘的伽师瓜。
1.2 主要试剂和试剂盒
Tween 20,国产分析纯;RNAplant Plus RNA提取试剂盒,TIANGEN BIOTECHCO公司产品;ReverTra Ace qPCR RT Kit,TOYOBO公司产品;SYBR○RGreen Realtime PCR MasterMix-Plus qRTPCR试剂,TOYOBO公司产品。
1.3 主要仪器设备
Centrifuge 5804R高速低温离心机,Eppendorf公司制造;SmartGel紫外凝胶成像系统,北京赛智创业科技有限公司制造;TP600 PCR仪,TakaRa公司制造;Personalcycler仪,Bionietra公司制造;HiSeqTM2000型测序平台,Illumina公司制造。
1.4 方法
1.4.1 样品处理与收集 青霉菌(Penicillium)孢子悬浮液配制:青霉菌分离自采后哈密瓜贮藏过程中发病的果实,经纯化培养后,用含质量分数0.01% Tween 20无菌水配制成浓度为1×105个/mL的孢子悬浮液,备用。将哈密瓜用质量分数0.05%的二氧化氯水消毒,再用无菌水清洗,沥干水分。用灭过菌的打孔器在哈密瓜赤道部位表面打6个深度为1~1.5 cm的孔,其中2个孔不接菌,其余4个孔中分别接入10 μL浓度为1×105个/mL的青霉菌孢子悬浮液。分别于0 h,接菌48 h(潜伏期)和60 h(显症期)取样。每组处理进行3个平行实验。取不接菌、接菌48 h、接菌60 h的哈密瓜组织样品备用;另取不接菌、接菌48 h、接菌60 h的哈密瓜组织样品进行混合,备用。
1.4.2 总RNA的提取 总RNA提取参照TIANGEN RNAplant Plus RNA提取试剂盒操作说明,并根据实际情况略作调整[14]。
1.4.3 转录组测序 转录组测序工作委托深圳华大公司完成。
1.4.4 转录组生物信息学分析
1)产量统计:测序得到的原始图像数据经base calling转化为序列数据,通过去除含 adaptor的reads,去除N的比率大于10%的reads,去除低质量reads(质量值Q≤10的碱基数占整个read的20%以上),获得Clean reads。
2)组装Contig和Unigene长度分布统计:用短reads组装软件Trinity组装,对组装出来的Contig 和Unigene做长度分布统计。
3)Unigene功能注释及 COG分类:分别将Unigene注释到NR(NCBI non-redundant),NT(NCBI nucleotide database),Swiss-Prot(the Swiss-Prot protein database),KEGG (the Kyoto Encyclopedia of Genes and Genomes),COG (the Clusters of Orthologous Groups of proteins),GO(gene ontology)数据库,对注释到每个数据库以及所有注释上的Unigene数目统计,给出NR分类图和COG分类图。
4)Unigene的GO分类:根据NR注释信息得到GO功能注释,得到每个Unigene的GO注释后,用WEGO软件对所有Unigene做GO功能分类统计。
5)Unigene代谢通路分析:根据KEGG数据库的注释信息,得到Unigene的Pathway注释。
2 结果与分析
2.1 产量统计
数据处理统计结果见表1。

表1 哈密瓜果实测序产量统计Table1 Output statistics of sequencing of Hami melon fruit
对哈密瓜果实测序后,共获57 872 626条原始reads片段,过滤后Clean Reads数为54 972 838条,含4 947 555 420 nt碱基数,其中过滤后Q20碱基比率为98.67%,过滤后不确定的碱基比率为0,GC值45.37%。可见,测序可满足后续数据组装及处理的要求。
2.2 组装Contig和Unigene长度分布统计
使用组装软件Trinity对 54 972 838条Clean Reads进行组装,获得了83 968条Contig片段,总长33 408 705 nt;获得了50 502条Unigene,总长41 882 740 nt,其中聚类的Unigene15 076条,单独的gene 35 426条。
结果见表2。
2.2.1 哈密瓜果实转录组的Contig长度分布及质量统计 对组装出来的Contig做长度分布统计。结果如图1所示。
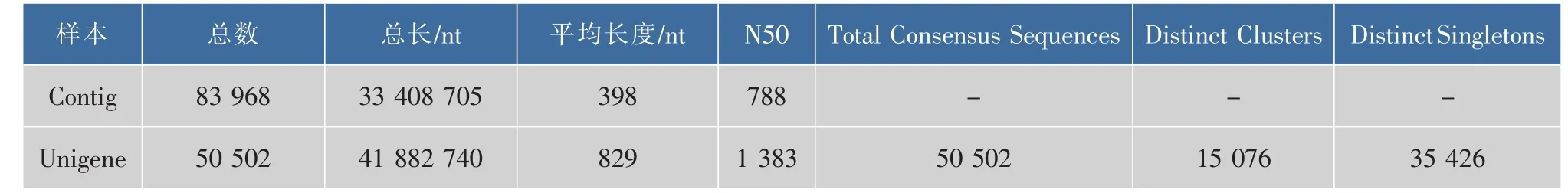
表2 哈密瓜果实转录组组装质量统计Table2 Statistics of assembly quality in transcriptome of Hami melon fruit
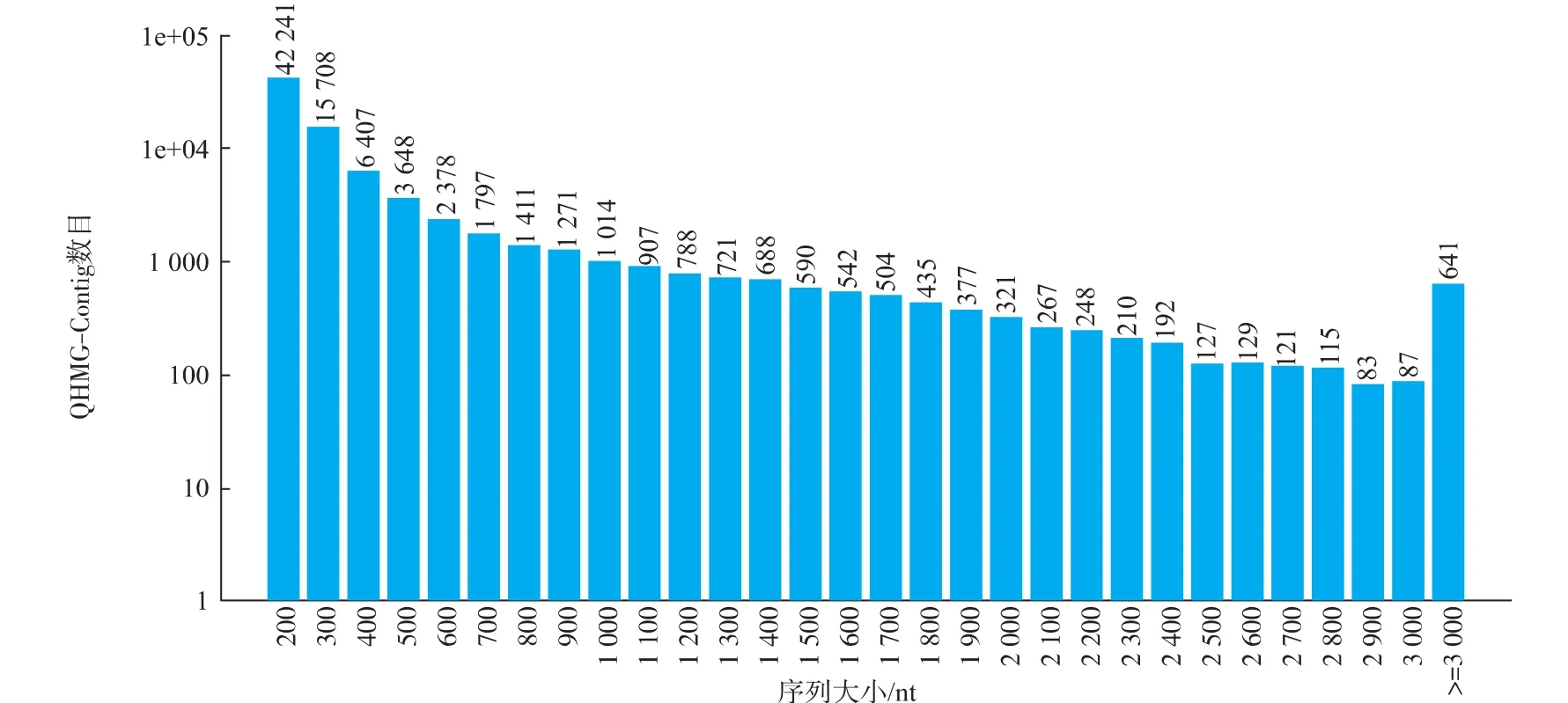
图1 哈密瓜果实转录组的Contig长度分布统计Fig.1 Length distribution of assembly Contigs in transcriptome of Hami melon fruit
组装出的83 968条Contig片段中,长度在100~200 nt的有42 241条,占50.31%;200~300 nt 的15 708条,占18.71%;300~400 nt的6 407条,占7.63%;400~500 nt的3 648条,占4.34%;大于等于500 nt的15 964条,占19.01%。所有Contigs总长为33 408 705 nt,平均长度398 nt,N50为788 nt。可见,Contig片段长度以100~200 nt为主,符合Illumina测序的预期结果。
2.2.2 哈密瓜果实转录组的Unigene长度分布及质量统计 对组装出来的Unigene做长度分布统计。由图2可知:组装出的50 502条Unigene中,100~500 nt的 Unigenes 25 837条,占总 Unigenes的 51.16%;500~1000nt的Unigenes10605条,占21.00%;1 000~1 500 nt的Unigenes 5 824条,占11.53%;1 500~2 000 nt的Unigenes 3 778条,占7.48%;大于等于2 000 nt的Unigenes 4 458条,占8.83%。所有Unigenes总长为41 882 740 nt,平均长度为829 nt,N50为 1 383 nt。在所有的 Unigenes中 Distinct Clusters有15 076条,Distinct Singletons有35 426条。对测序获得的read在组装好的Unigene的位置分布情况进行分析,结果如图3所示。哈密瓜果实转录组测序获得的Unigene 3'端和5'端的reads数量相对较少,尤其是3'末端和5'末端位置,而相对位置在0.2~0.8的部分,reads数量相对较多且分布较均衡。
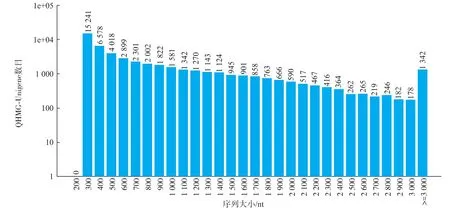
图2 哈密瓜果实转录组的Unigene长度分布统计Fig.2 Length distribution of assembly Unigene in transcriptome of Hami melon fruit
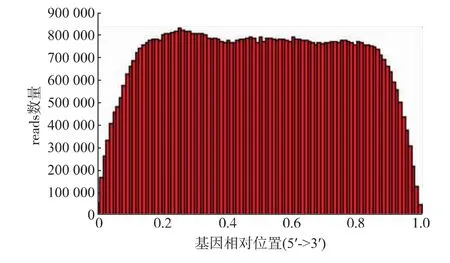
图3 哈密瓜果实文库的reads在Unigene上的测序随机性分布Fig.3 Randomicity of Hami melon fruit reads on Unigene
2.3 哈密瓜果实转录组的Unigenes功能注释及COG分类
以多种互补方法对拼接的Unigenes作功能注释。通过blastx将Unigene序列比对到蛋白质数据库 NR、Swiss-Prot、KEGG和 CO G (evalue<0.000 01),并通过blastn将Unigene比对到核酸数据库Nt(evalue<0.000 01),得到跟给定Unigene具有最高序列相似性的蛋白质,从而得到该Unigene的蛋白质功能注释信息。
2.3.1 注释结果汇总 统计结果见表3:在50 502条Unigene中,36 000条Unigene(71.28%)、37 256条Unigene(74.31%)和21 002条Unigene(41.59%)在设定的E值范围内可以比对到NR数据库、NT数据库和Swiss-Prot数据库,但还有28.72%的Unigene 在NR库中是未知基因,25.69%的Unigene在NT库中是未知序列,29 500条Unigenes(58.41%)在Swiss-Prot数据库中没有得到注释。将50 502条Unigenes比对到 KEGG数据库,有 19 600条Unigenes参与了代谢通路;有13 221条Unigenes在COG数据库可以预测功能;有27 001条Unigenes可以映射到GO不同的功能节点上。
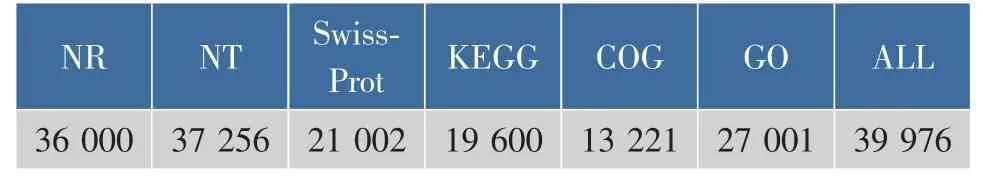
表3 注释结果统计Table3 Statistics of annotation results
没有比对到 NR、NT和 Swiss-Prot数据库的10 526条Unigene可能是新发现的转录本,是哈密瓜果实区别于其它物种而特有的基因,这需要进一步验证。
2.3.2 哈密瓜果实转录组的Unigene在NR数据库中的注释分类结果 在NR数据库中,以E-value值10-5为截点进行 BLAST比对,共有 71.28%的Unigenes比对到该数据库中。它们中 38.62%的Unigenes E-value值低于1E-100,25.28%的Unigenes E-value值在 1E-100与 1E-45之间,36.12%的Unigenes E-value值在1E-45与1E-5之间,详见图4(a),表明注释到NR数据库的Unigenes与库内基因具有很好的同源性。相似度分析显示,在能比对到Nr数据库的Unigene中,相似度在20%~40%的Unigene为1 490条,占总Unigene的4.14%;相似度在 40%~60%的 Unigene为 2 370条,占总Unigene的6.58%;相似度在60%~80%的Unigene 为2 891条,占总Unigene的8.03%;相似度在80%~95%的 Unigene为 11 754条,占总 Unigene的32.65%;相似度在95%~100%的Unigene为17 495条,占总Unigene的48.60%,见图4(b)。图4(c)为注释到NR数据库中Unigene匹配序列的种群分布图,可见,样品注释到NR数据库的Unigene与黄瓜已发表基因组序列匹配度最高,达85.61%;其次是甜瓜,2.54%;葡萄1.54%;桃1.24%。Unigene数目分别为30 820条、916条、556条、445条。
2.3.3 哈密瓜果实转录组的Unigene COG功能分类 哈密瓜果实转录组中Unigene有23 421条与COG数据库中的基因建立了对应关系,根据功能可将Unigene分为25类,分别用A—Z表示,见图5。Unigene的COG功能涉及大多数的生命活动,其中,R类 General function prediction only基因数量最多,为3 946条,占16.85%;其次是L类Replication,recombination和repair基因数量2150条,占 9.18%;K类 Transcription基因数2 043条,占8.72%;O类 Posttranslational modification,protein turnover,chaperones基因数量1 796条,占7.67%;J类Translation,ribosomal structure和biogenesis基因数量1 670条,占7.13%;T类Signal transduction mechanisms基因数量为1 581条,占6.75%;G类Carbohydrate transport和 metabolism 基因数量为1 405条,占6.00%;E类 Amino acid transport和metabolism基因数量 1 014条,占 4.32%;W 类Extracellular structures和Y类Nuclear structure基因数量最少,仅为9条和5条。

图4 NR分类结果Fig.4 Figure of NR classification
2.4 哈密瓜果实转录组的Unigene GO功能分类
基于NR注释进行GO分类,经BLAST2G0软件分析,发现在39 976个注释序列中有27 001个能得到相应的GO注释。通过WEGO软件分类统计,这些GO信息被进一步归纳为基因的分子功能、细胞组分、生物过程3个主类,54个亚类,具体见图6。
由于同一转录本可映射到不同节点,所以共有83 951条Unigene归入细胞组分,主要聚集于细胞、细胞成分和细胞器3个亚类,分别有21 254、21 253 和17023个Unigene归类于此3个亚类;有30788条Unigene归入分子功能,主要聚集于结合作用和催化活性两个亚类,分别有 13 057和 13 118个Unigene被归类于此 2个亚类;有110 204条Unigene归入生物学过程,生物过程中样品基因主要聚集于细胞过程、代谢过程和单个有机体过程3个亚类,分别有17 329、16 505和12 142个Unigenes被归类于此3个亚类;其它涉及生物调节的Unigene有6 949条,涉及应激反应的有8 585条,涉及信号的Unigene有2 442条。表明这些功能在哈密瓜抗病过程中起着重要作用。
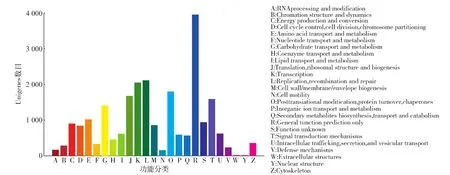
图5 COG功能注释分布Fig.5 COG function classification of Unigene in All-Unigene
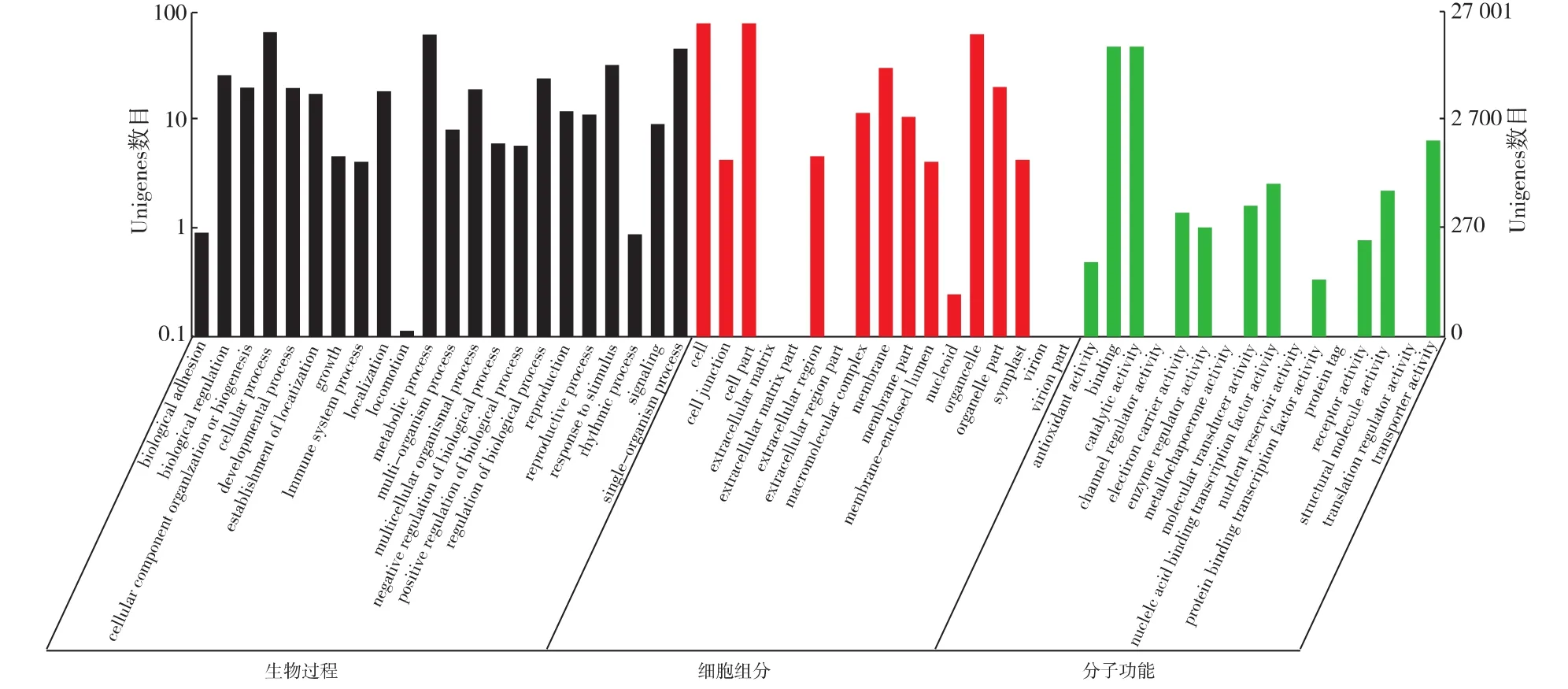
图6 Unigene的GO分类Fig.6 GO function classification of Unigene in All-Unigene.
2.5 哈密瓜果实转录组Unigene代谢通路分析
2.5.1 哈密瓜果实转录组Unigene代谢通路分析为系统分析基因产物在细胞中的代谢途径以及这些基因产物的功能,将本次转录组测序得到的39 976个 Unigene比对到 KEGG数据库进行Pathway注释。结果发现有19 600个(49.0%)成功获得注释,共映射到不同的代谢通路128个。包括生化代谢通路、植物激素信号转导、植物-病原互作、RNA转运、甘油磷脂代谢、糖酵解、糖异生、苯丙氨酸生物合成、核苷酸剪切修复、脂类代谢、嘌呤代谢、类固醇生物合成等,具体详见文献[15]。
在不同的代谢通路中涉及到的Unigenes数量差异较大,其中代谢途径的Unigene最多,为4 388个(22.39%),其次是次生代谢产物的生物合成相关的Unigene,有2 031个(10.36%),植物激素信号传导相关的Unigene有965个(4.92%),植物-病原物互作相关的Unigene有741个(3.78%),而参与甜菜素生物合成的Unigene最少,仅为1个(0.01%)。
对哈密瓜果实转录组中与环境适应性、免疫系统、信号转导相关的Unigene进一步分析,由图7、8、9可知,与环境适应性相关的Unigene有971条,其中与植物—病原菌互作的Unigene有741条;与免疫系统相关的Unigene有116条;与信号转导相关的Unigene有1 038条。这些Unigene功能的挖掘,将为采后哈密瓜对外界逆境的分子应答机制奠定基础。
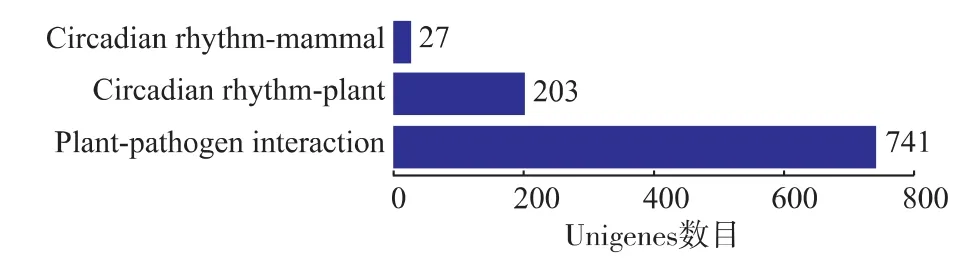
图7 代谢途径中与环境适应相关的UnigeneFig.7 Unigene related to environmental adaptation in metabolic pathways
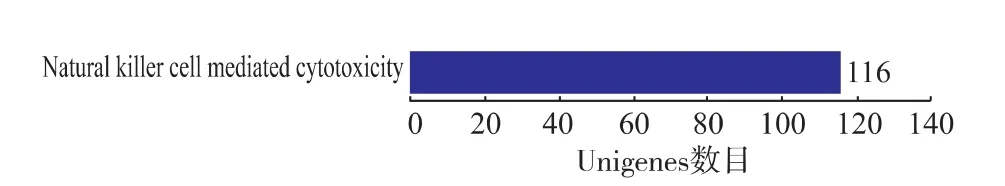
图8 代谢途径中与免疫系统相关的UnigeneFig.8 Unigene related to immune system in metabolic pathways
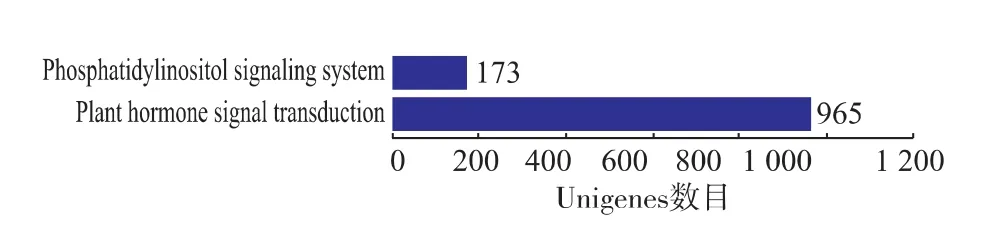
图9 代谢途径中与信号转导相关的UnigeneFig.9 Unigene related to Signal transduction in metabolic pathways
2.5.2 哈密瓜果实转录组植物与病原菌相互作用通路分析 针对哈密瓜抗病性的研究,植物-病原菌互作代谢通路成为关注重点,见图10。进一步分析可知,植物-病原菌互作的Pathway通路中含有fungal PAMP,Ca2+,Bacterial flagellin,Bacterial EFTu,Bacterial secretion system,PRRs,Coronatine/JA,Defense-realated gene induction等子代谢通路,其基因数目见表4。

图10 哈密瓜果实中植物与病原菌相互作用途径KEGG预测图Fig.1 0 Plant-pathogen interaction pathway in Hami melon fruit generated by KEGG
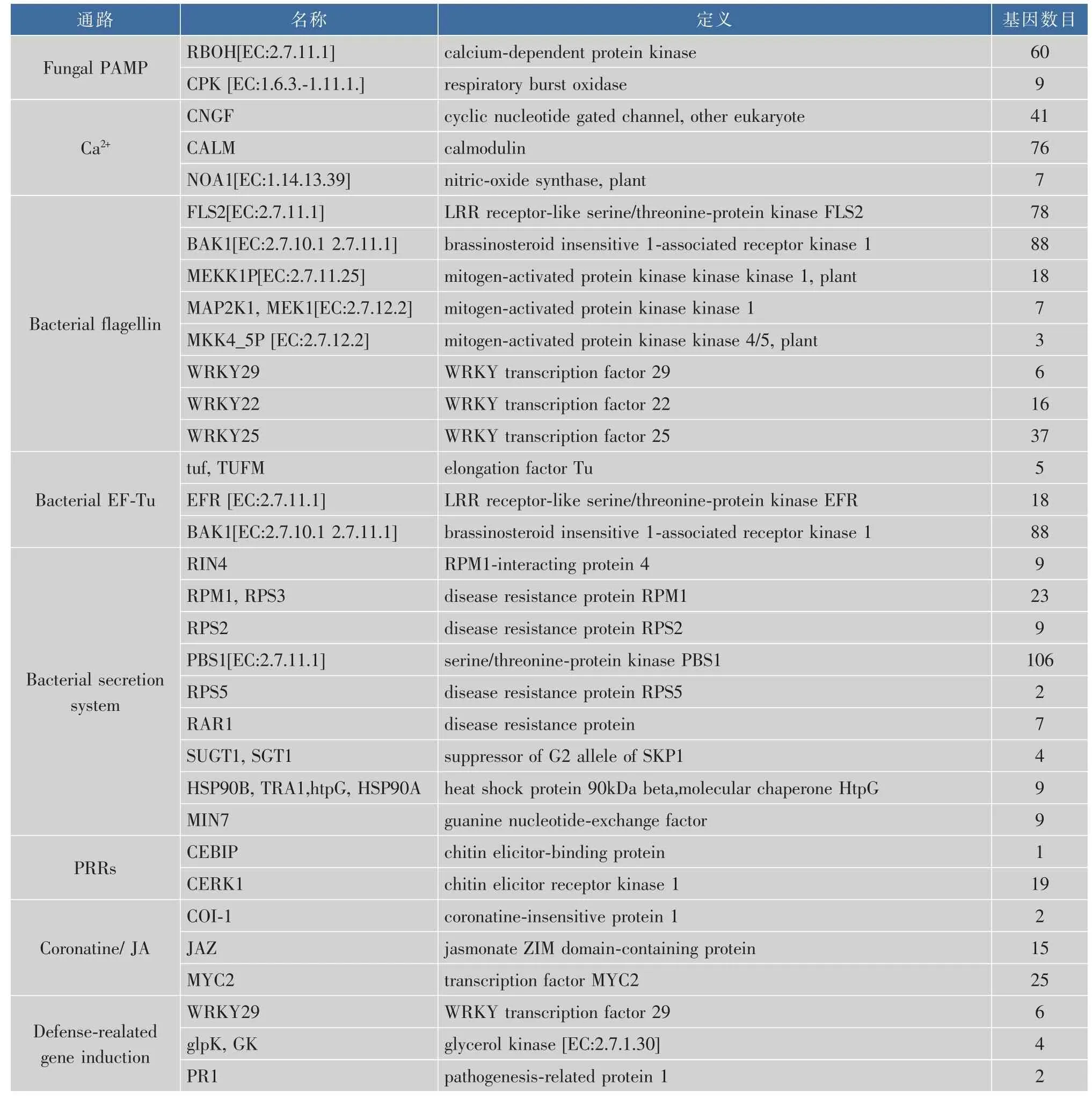
表4 哈密瓜果实中植物与病原菌相互作用相关基因Table4 Plant-pathogen interaction pathway related genes in Hami melon fruit
3 结语
利用Illumina平台测序并分析哈密瓜的转录组信息,获得过滤后的Clean Reads数为54 972 838条,包含4 947 555 420 nt的碱基数,使用组装软件Trinity对54 972 838条Clean Reads进行组装,获得了50 502条Unigene,总长41 882 740 nt。在NR数据库中,以E-value值10-5为截点进行BLAST比对,共有71.28%的Unigenes比对到该数据库中,注释到NR数据库中的Unigene与黄瓜的已发表基因组序列匹配度最高,达85.61%。但还有10 526条Unigene没有比对到NR、NT和Swiss-Prot数据库,可能是新发现的转录本,是哈密瓜果实区别于其它物种的特有基因,这需进一步验证。为系统分析基因产物在细胞中的代谢途径,以及这些基因产物的功能,将本次转录组测序得到的39 976个Unigene比对到KEGG数据库进行Pathway注释。结果发现,有19 600个(49.0%)成功获得注释,共映射到不同的代谢通路128个。目前国内还没有关于新疆哈密瓜转录组学的数据库信息。本实验结果为采后哈密瓜的转录组学分析研究提供了一种切实有效的模型,为开展相关抗病性基因的表达调控奠定了基础,进而为哈密瓜采后病害的无污染防治技术和绿色保鲜技术提供了理论依据。
[1]杨渡.浅谈新疆甜瓜产业发展[J].新疆农业科学,2002,39(1):1-5. YANG Du.Elementary introduction to development of muskmelon industry of Xinjiang[J].Xinjiang Agricultural Sciences,2002,39(1):1-5.(in Chinese)
[2]张辉,李学文.新疆哈密瓜果实潜伏侵染真菌种类研究[J].新疆农科院园艺所,2002(3):127-130. ZHANG Hui,LI Xuewen.Study on the species of latent fungi of Xinjiang Hami melon fruit[J].Xinjiang Agricultural Academy of Sciences,2002(3):127-130.(in Chinese)
[3]赵劼.厚皮甜瓜主要采后病害的潜伏侵染及生物学控制[D].兰州:甘肃农业大学,2003.
[4]毛晓英,吴庆智,刘晓航,等.新疆哈密瓜采后主要致腐病原真菌的分离与鉴定[J].安徽农学通报,2006,12(12):120-121. MAO Xiaoying,WU Qingzhi,LIU Xiaohang,et al.Separation and identification of pathogenic fungi from the postharvest Hami melon[J].Anhui Agri Sci Bull,2006,12(12):120-121.(in Chinese)
[5]邓志斌,郭图强,侯建华,等.新疆哈密瓜生产存在的问题与对策[J].新疆农垦科技,2003(6):15-16. DENG Zhibin,GUO Tuqiang,HOU Jianhua,et al.Xinjiang Hami melon production problems and counter measures[J].Xinjiang Agricultural Reclamation Technology,2003(6):15-16.(in Chinese)
[6]STURDEVANT D E,VIRTANEVA K,MARTENS C,et al.Host-microbe interaction systems biology:life cycle transcriptomics and comparative genomics[J].Future Microbiol,2010,5(2):205-219.
[7]WANG Z,GERSTEIN M,SNYDER M.RNA-Seq:a revolutionary tool for transcriptomics[J].Nat Rev Genet,2009(10):57-63.
[8]FAUNES F,SANCHEZ N,CASTELLANOS J,et al.Identification of novel transcripts with differential dorso-ventral expression in xenopus gastrula using serial analysis of gene expression[J].Genome Biol,2009(10):15.
[9]YASSOURA M,KAPLANA T,FRASER H B,et al.Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing[J].Proc Natl Acad Sci USA,2009,106:3264-3269.
[10]XIE C J,WANG D,YANG X Y.Protein extraction methods compatible with proteomic analysis for the cotton seedling[J].Crop Science,2009,49:395-402.
[11]WANG Xiaojie,LIU Wei,CHEN Xianming,et al.Differential gene expression in incompatible interaction between wheat and stripe rust fungus revealed by the cDNA-AFLP and comparison to compatible interaction[J].BMC Plant Biology,2010(10):1-15.
[12]LEROCH Michaela,KLEBER Astrid,SILVA Evelyn.Transcriptome profiling of botrytis cinerea conidial germination reveals upregulation of infection-related genes during the prepenetration stage[J].Eukaryot Cell,2013,12(4):614-626.
[13]ZABALA Gracia,ZOU Jijun,TUTEJA Jigyasa.Transcriptome changes in the phenylpropanoid pathway of glycine max in response to pseudomonas syringae infection[J].BMC Plant Biology,2006(6):26.
[14]MARIONI J C,MASON C E,MANE S M,et al.RNA-seq:an assessment of technical reproducibility and comparison with gene expression arrays[J].Genome Res,2008,18:1509-1517.
[15]单春会.哈密瓜响应青霉菌侵染的转录组和蛋白质组研究及相关抗性酶变化分析 [D].无锡:江南大学食品学院,2015:19-22.
Construction and Analysis of the Transcriptome Before and After Infected by Penicillium in Hami Melon Fruit
SHAN Chunhui1,2, CHEN Wei*1, TANG Fengxian2, JIANG Fuyao2
(1.School of Food Science and Technology,Jiangnan University,Wuxi 214122,China;2.Food College,Shihezi University,Shihezi 832003,China)
Hami melon is famous with its unique flavor and rich nutrition.However,it is easily spoilt due to infected by Penicillium.The changes of transcriptome levels before and after infected by Penicillium in Hami melon were investigated by illumina paired-end sequencing.The results showed that the filtrated clean reads contained 4 947 555 420 nt nucleotides,further assembling the Clean Reads led to the product of 50 502 unigenes.A total of 71.28%unigenes were found in NR database using BLASTX comparison with an E-value cut-off of 1e-5,whereas 10 526 unigenes were still found to no significant matches to any known protein in NR,NT and Swiss-Prot database.These may be the specific genes leading to the difference between Hami melon and other species and need to be further verified.The unigene metabolic pathway analysis was conducted on the 39 976 unigenes through comparing to KEGG database and 128 metabolic pathways were mapped.Transcriptional analysis will greatly improve the understanding of postharvest physiology characteristics of Hamimelon at the molecular level.It will provide a practical and effective model for the future research,such asexploringtheexpressionandregulationofimportantgenesrelatedtoPenicilliumpathogenresistance.
Hami cantaoupe,Transcriptome,De novo,Assembly analysis
Q 786
A
1673—1689(2016)06—0806—09
2015-01-27
国家自然科学基金项目(31360412)。
单春会(1978—),男,新疆石河子人,工学博士,副教授,主要从事食品生物技术的研究。E-mail:792875996@qq.com
陈 卫(1966—),男,江苏扬州人,工学博士,教授,博士研究生导师,主要从事食品生物技术的研究。E-mail:sch_0909@163.com
