姜黄素的结构修饰与抗肿瘤研究进展
2016-09-14李家庆长江职业学院生物医药学院湖北武汉430064
李家庆(长江职业学院生物医药学院,湖北 武汉 430064)
姜黄素的结构修饰与抗肿瘤研究进展
李家庆
(长江职业学院生物医药学院,湖北 武汉 430064)
姜黄素是一种天然的多酚类物质,广泛分布在多种植物中,并具有多种生物活性。但是其存在自身抗肿瘤作用较弱、生物利用度低等缺点,使其临床应用受到了限制。基于此,研究人员对姜黄素进行了大量的结构修饰,以期改善其抗肿瘤活性和成药性。本研究主要对姜黄素的结构修饰及抗肿瘤作用研究进展进行综述,旨在为以后合成高效专一活性的姜黄素衍生物提供依据。
姜黄素;结构修饰;抗肿瘤活性
姜黄素是一种天然的多酚类物质,广泛分布在姜黄属植物中如姜黄、莪术、郁金等,具有抗癌、抗炎、抗氧化、降血脂等多种药理学活性[1],而且对正常细胞基本没有毒性。作为抗肿瘤活性成分,姜黄素不仅能够诱导肿瘤的分化及凋亡,抑制血管的生成,而且还可以调节抑癌基因、癌基因蛋白的表达[2],有望成为一类高效低毒的天然新型抗肿瘤药物。然而,姜黄素也存在一些缺点如自身抗肿瘤作用较弱、生物利用度低、水溶性差等,这些都限制了其在临床上的应用。因此,国内外的研究者们对姜黄素进行大量的结构修饰来提高姜黄素的抗肿瘤活性,并改善其成药性质。
从结构上看,姜黄素是一类1,7-二( 4-羟基-3-甲氧基)苯基-1,6-庚二烯-3,5-二酮类化合物 (图1),研究人员对其结构的改造主要是集中在芳香环和连接桥链上,由此获得了一些具有靶向性、高活性的衍生物。本文主要对近年来姜黄素结构修饰及抗肿瘤作用的研究进展进行综述,旨在为其进一步开发姜黄素衍生物提供参考。
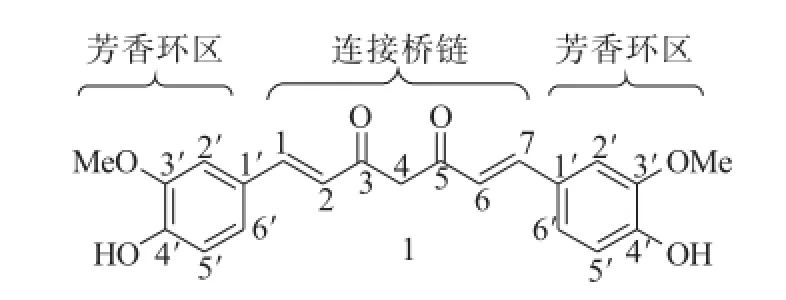
图1 姜黄素
1 苯环的修饰
对姜黄素苯环的修饰主要包括将酚羟基成醚、成酯、增加或改变酚羟基的位置、去掉酚羟基或甲氧基以及在苯环上引入其他基团等修饰。
1.1苯环的酚羟基成醚或酯
Shi等[3]将姜黄素的酚羟基修饰成甲氧基得到化合物2,对前列腺癌PC-3和LNCaP展现出了较强的抑制活性,其IC50分别为1.1mM和1.3mM。进一步研究发现,化合物2可能是通过雄激素的降解作用来发挥抗肿瘤活性。
除了成醚外,将酚羟基修饰成酯也能够提高抗肿瘤活性。Mishra等[4]将姜黄素的酚羟基修饰成不同的酯,其中化合物3~4通过下调与凋亡相关的蛋白Bcl-2等,诱导AK-5肿瘤细胞的凋亡。
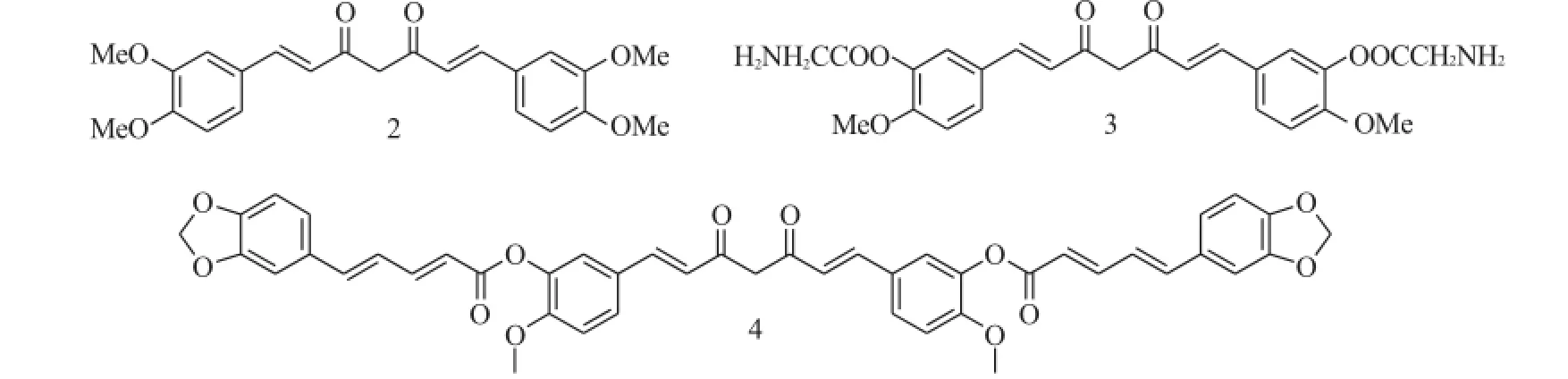
图2 化合物2~4
1.2苯环增加或改变酚羟基的位置、去掉酚羟基或甲氧基
Venkateswarlu等[5]合成了一系列的多羟基类姜黄素衍生物,活性测试发现,这类多羟基化合物具有较好的清除过氧基化合物和DPPH自由基的作用,如化合物5对道尔顿淋巴瘤有较强的抑制活性。
另外,Ishida等[6]研究发现,用氟原子代替姜黄素的酚羟基 (化合物6)对多种肿瘤细胞具有较强的抑制作用。
有鉴于此,本文试图挖掘《孤独的割麦女》一直以来被忽略的“权力事实”[7],探寻诗歌中所暗含的政治涵义。本文就诗人的凝视行为与割麦女吟唱行为的交锋以及歌声中的战争与文化冲突来展开讨论。
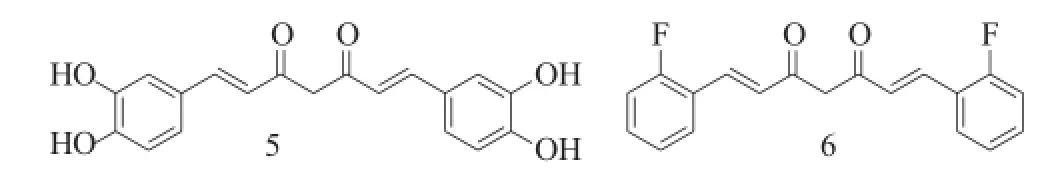
图3 化合物5~6
1.3苯环被甲基或杂环取代
Lin等[7]发现,将姜黄素的一个苯环用甲基取代后得到的化合物7也具有较强的抗肿瘤活性。但是,将苯环用五元杂环(化合物8~9)取代后降低了抗癌活性。

图4 化合物7~9
2 1,6-庚二烯-3,5-二酮连接的修饰
2.1活性亚甲基的修饰
由于姜黄素的连接链是一个大共轭体系以及b-二酮的结构,因此姜黄素的亚甲基有较高的活性并有一定的酸性,能够被修饰。事实上,目前对姜黄素的修饰主要是集中在亚甲基上。
Ohtsu等[8]以姜黄素为先导化合物,主要是在C4位亚甲基引入一系列的取代基,其中以引入丙酸乙酯(化合物10)具有较强的抗前列腺癌的活性。但是将丙酸乙酯修饰成丙烯酸乙酯(化合物11)、丙烯氰(化合物12)等均降低了化合物抗前列腺癌的活性[7]。
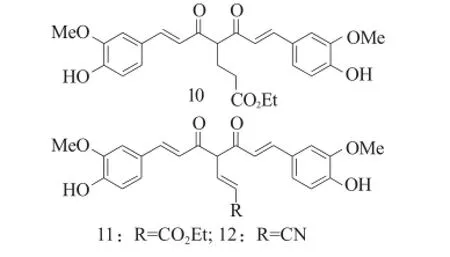
图5 化合物10~12
除了引入丙烯酸甲酯基能够提高姜黄素的抗肿瘤外,在姜黄素的C4位引入苯甲烯结构也能够显著提高抗癌活性,如化合物13~16对肺癌A549的抑制活性明显高于姜黄素,其GI50在0.35~0.55mM[9]。
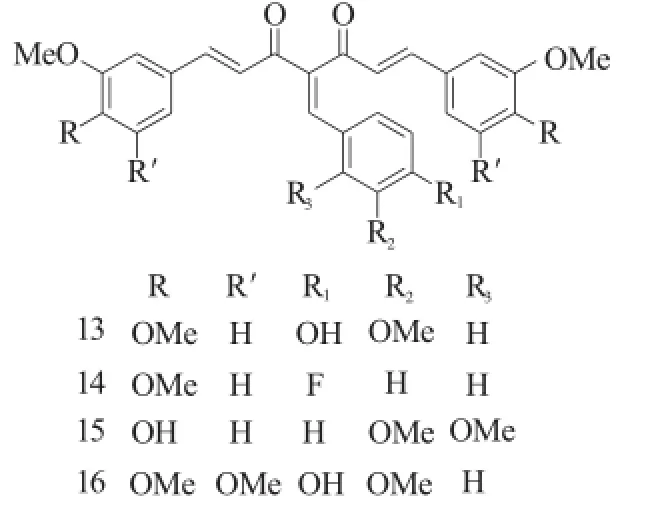
图6 化合物13~16
图7 化合物17
2.2单羰基的修饰
将姜黄素连接链修饰成单羰基也是姜黄素结构修饰的一个研究热点,其中部分单羰基姜黄素衍生物也展现出了优秀的抗肿瘤活性。
Fuchs等[11]将姜黄素的2个羰基去掉一个后得到的单羰基衍生物对前列腺癌PC-3、LNCaP细胞和乳腺癌MCF-7、MDA-MB-231细胞的抑制活性均强于姜黄素,其中化合物18的活性最强,其对4种肿瘤细胞的IC50分别是2.1μM、0.5μM、0.4μM、0.6μM。Zhou等[12]以化合物18为先导化合物,将其一个苯环用环己烯取代得到了一系列的衍生物,这些衍生物 (化合物19)只展现出了中等的抗前列腺癌活性。
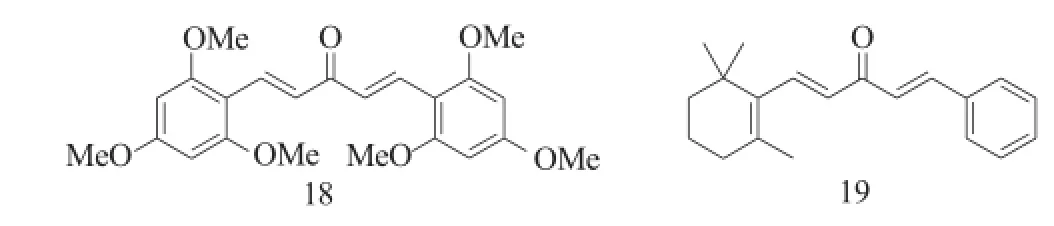
图8 化合物18~19
Liang等[13]也是以化合物18为先导化合物,在C4位引入环戊烷或环己烷。其中环戊烷的衍生物对KB和LeLa细胞有着较好的抑制作用,如化合物19 (IC50<9.3μM)对KB的抑制作用最强,是姜黄素 (IC50=35.9μM)的4倍。相比环戊烷,环己烷类化合物对HL-60和KB有着较强的抑制活性,其中化合物20对KB (IC50=6.7μM)的抑制作用最强,是姜黄素的5倍。Suarez等[14]也是以化合物18为先导化合物,将其羰基用丙二氰取代得到了化合物21,该化合物对多种肿瘤细胞均有较强的抑制活性,其中对NCI-ADR的抑制活性最强,其IC50值为2.97μM。
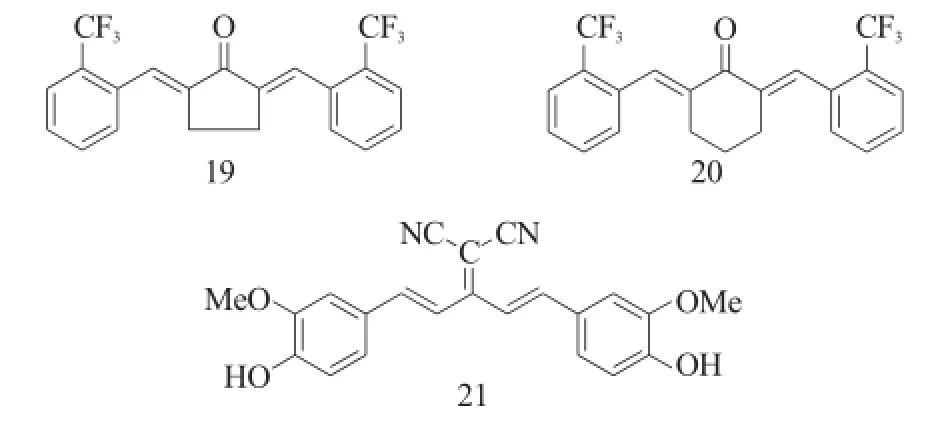
图9 化合物19~21
Tan等[15]则以化合物19为先导化合物,将环戊烷修饰成哌啶环或噻己烷。总的来说,环戊烷类化合物的抗肿瘤活性比环己环、哌啶环和噻己环类化合物的活性低;而哌啶环和噻己环类化合物对HL-60和K562的抑制活性又明显高于环己环类化合物,但是将环己环类化合物上的一个苯环用吡啶环取代后抗肿瘤活性显著增加了,其中化合物22对NB4、NB4-R1、HL-60、K562等的抑制活性最强,其IC50值分别是0.19μM、0.24μM、0.18μM、0.24μM。
Wei等[16]则基于杂环类似化合物能够提高抗肿瘤活性,合成了一系列的哌啶、吡喃和噻己环类的化合物,其中吡喃类化合物的抗肿瘤活性最强,如化合物23对PC-3、Panc-1和HT-29的抑制活性IC50分别是0.27μM、0.52μM、0.16μM。
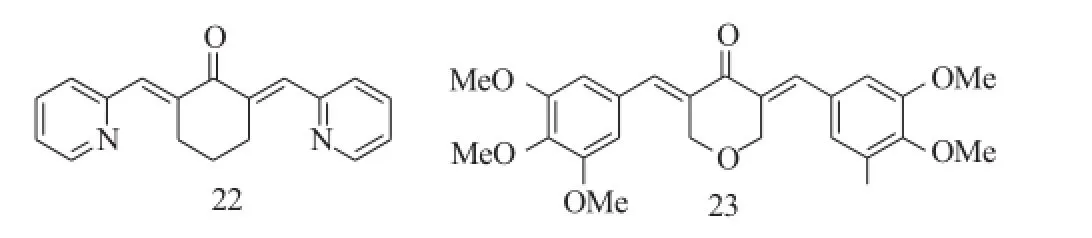
图10 化合物22~23
Yadav等[17]则在姜黄素C4位引入哌啶环并将氮原子进行了甲基化,这种含有哌啶环的化合物比含有环戊烷或环己烷的化合物具有更强的抗乳腺癌活性,其中化合物24对乳腺癌MDA-MB-231的抑制活性最强,其EC50为0.3μM。Simoni等[18]也发现哌啶类化合物25对于Caco-2和SH-SY5Y有着较强的抑制活性,然而该化合物对正常的细胞也有较强的毒性。但是,在化合物25哌啶基的氮原子上引入一系列的具有神经保护的基团,能明显降低对正常细胞的毒害作用,并保留较强的抗肿瘤活性 (化合物26。Makarov等[19]合成了一系列的苯环被噻吩环取代的哌啶磷酰胺类化合物,这些化合物也对多种肿瘤细胞有着较强的抑制活性,其中化合物27对Scov-3、Caov-3和A549的抑制活性最强,其IC50分别为8μM、10μM、4μM。Kalai等[20]也对哌啶氮进行衍生化,主要是将一系列的吡咯环引入到哌啶氮上,同时在苯环上用氟或三氟甲基进行取代。这些含吡咯环的化合物对A2780,MCF-7和H9c2均展现出了较好的抑制活性,其中化合物28对这3种肿瘤细胞的抑制活性最强。
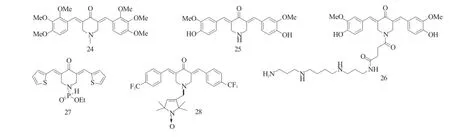
图11 化合物24~28
Das等[21]则以化合物24为先导化合物,通过一系列的连接片段将2个化合物23的类似物进行拼接得到了一系列的二聚化合物29,这些化合物对HSC-2、HSC-3、HSC-4、HL-60均有较强的抑制活性,其中化合物29b的抑制活性最强,其IC50值分别为0.050μM、0.046μM、0.084 μM和0.064μM。但是,在化合物29b的苯环上引入取代基降低了抗癌活性[22]。Hela等[23]则通过连接片段拼接了一系列的含碱性侧链 (化合物30和31),以模仿抗乳腺癌药物他莫昔芬。这些碱性侧链的引入提高了化合物 30 (IC50=0.65μM)、31 (IC50=0.49μM)对 HCT-116的抑制活性,其活性至少是阳性药物5-氟尿嘧啶(IC50=3.56 μM)的5倍。
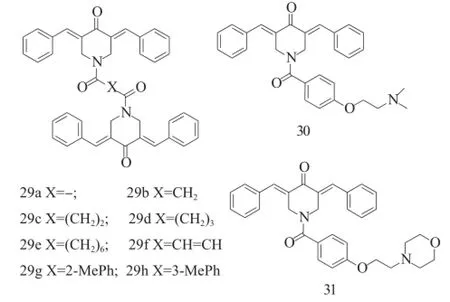
图12 化合物29~31
Zhang等[24]则以姜黄素为先导化合物,将苯环上的酚羟基进行修饰,一系列的含氮杂环 (咪唑、吲哚)被引入到了酚羟基上得到了一系列的衍生物。总的来说,这些含氮杂原子的引入(如化合物32)并没有增加抗肿瘤活性,反而丧失了对一些肿瘤细胞的抑制活性。但是将含氮的杂环改为酰胺后却能保留对多种肿瘤细胞的抑制活性,其中化合物33对多种肿瘤细胞的抑制活性甚至略高于先导化合物姜黄素。
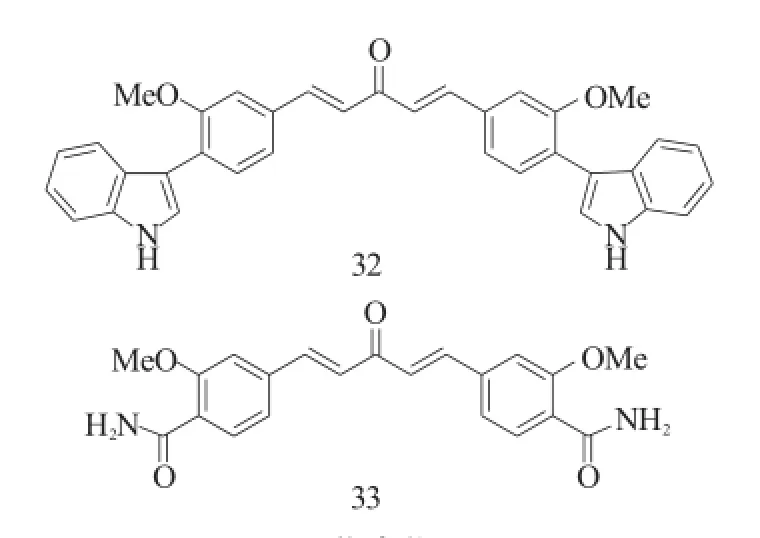
图13 化合物32~33
Woo等[25]则将苯并咪唑环取代姜黄素的苯环,这些苯并咪唑化合物对MCF-7、SH-SY5Y、HepG2 和H460的抑制活性均强于姜黄素,其中化合物34对这4种肿瘤细胞的抑制活性最强,其IC50分别是1.9μM、4.6μM、3.7μM、7.2μM。但是与他莫昔芬相比这些化合物的抗肿瘤活性仍然较弱。

图14 化合物34
2.3b-二酮的修饰
姜黄素具有b-二酮结构,酮基能够参与缩合等反应来构建一系列杂环化合物。Simoni等[26]将姜黄素的一个羰基用氨基取代,其中脂肪胺化合物35a、35c和35e展现了中等抗乳腺癌的活性,而含有芳香胺的衍生物(化合物35b和35d)却没有抗癌活性。当2个羰基被转化成异唑(化合物36,IC50=13.1μM)后,抗乳腺癌MCF-7的活性是姜黄素(IC50=29.3μM)的2倍。但是,当羰基被转化成肟衍生物时,却是含有苯环的肟(化合物37a,IC50=7.1μM)能够增强抗乳腺癌活性,而烷烃取代(化合物37b,IC50=36.3μM)却降低活性。
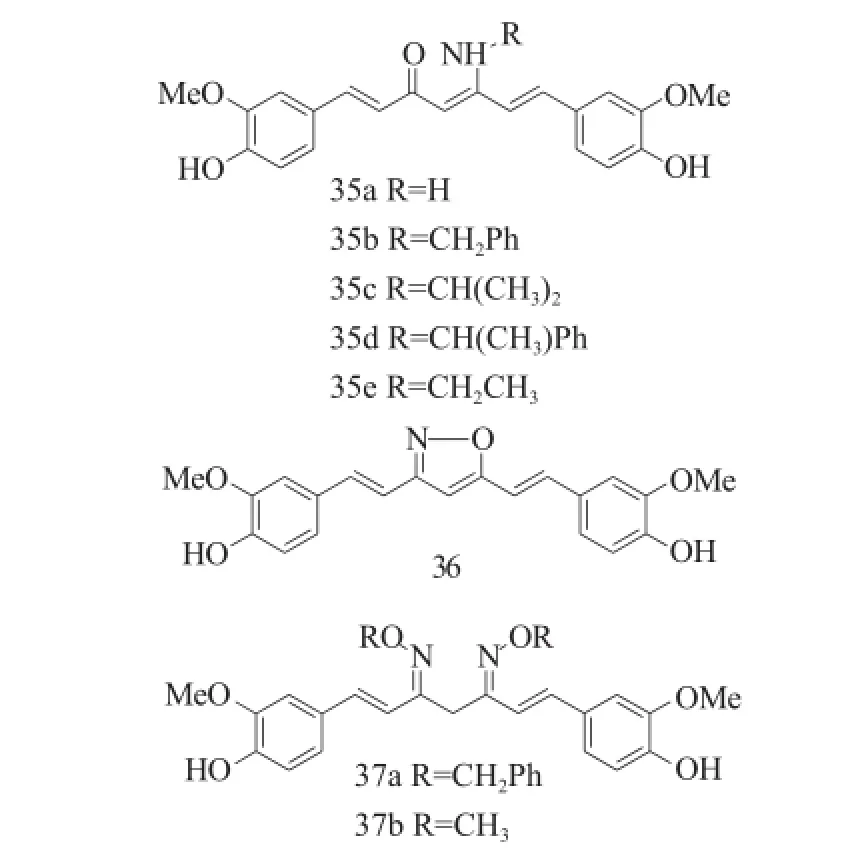
图15 化合物35~37
Ishida等将姜黄素的2个羰基与肼反应得到了咪唑类化合物38,该化合物对多种肿瘤细胞有着较强的抑制作用。此外,Qiu等[27]也以姜黄素为先导化合物,将b-二酮的2个羰基形成嘧啶环后增加抗肿瘤活性,其中化合物39展现出了最强的抗肿瘤活性,其抑制HT29和HCT-116的IC50值分别为7.1μM和6.2μM。
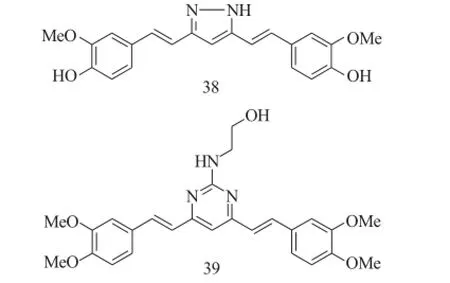
图16 化合物38~39
3 结语
本文对近年来姜黄素衍生物的研究进行总结,并初步阐述结构修饰对姜黄素抗肿瘤活性的影响,大致可以总结出以下几个规律:1)姜黄素的C4位酚羟基对抗肿瘤活性影响较大,将其修饰成醚、酯或用其他基团取代,能够增强抗肿瘤活性;2)姜黄素的C4亚甲基引入适当的取代基如苯甲烯基,能够提高抗肿瘤活性;3)姜黄素的b-二酮转化为单羰基后,包括含有杂环的单羰基或单羰基二聚物均能提高抗肿瘤活性;4)姜黄素的b-二酮结构也是一个抗肿瘤药效团,而该药效团能够被吡唑、嘧啶等杂环取代并保留抗肿瘤活性,对一些肿瘤细胞的抑制活性甚至有提高。
总之,对姜黄素进行结构修饰能够提高抗肿瘤活性,并取得一定的进展,但是目前报道的姜黄素衍生物抗癌效果与上市的抗癌药物相比仍然较弱。因此,进一步对姜黄素进行结构修饰以筛选出生物活性高效、专一的姜黄素衍生物值得研究。
[1] 郭晓丹,许建华.姜黄素及其衍生物抗肿瘤作用的研究进展[J].海峡药学,2011,23(6):15-18.
[2] 刘红艳,王海燕,叶松.姜黄素药理作用及其机制研究进展[J].中国现代医药杂志,2012,22(6):48-51.
[3] Shi Q, Shih C, Lee H. Novel anti-prostate cancer curcumin analogues that enhance androgen receptor degradation activity [J]. Anticancer Agents Med Chem, 2009(8): 904-912.
[4] Mishra S, Kapoor N, Mubarak A, et al. Differential apoptotic and redox regulatory activities of curcumin and its derivatives [J]. Free Radic Biol Med, 2005, 38(10): 1353-1360.
[5] Venkateswarlu S, Ramachandra M S, Subbaraju G V. Synthesis and biological evaluation of polyhydroxycurcuminoids [J]. Bioorg Med Chem., 2005, 13(23): 6374-6380.
[6] Ishida J, Ohtsu H, Tachibana Y, et al. Antitumor agents. Part 214: synthesis and evaluation of curcumin analogues as cytotoxic agents [J]. Bioorg Med Chem, 2002(11): 3481-3487.
[7] Lin L, Shi Q, Nyarko A K, et al. Antitumor agents. 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents[J]. J Med Chem., 2006, 49(13):396-3972.
[8] Lin L, Shi Q, Su C Y, et al. Antitumor agents 247. New 4-ethoxycarbonylethyl curcumin analogs as potential antiandrogenic agents[J]. Bioorg. Med. Chem., 2006, 14(8):2527-2534.
[9] Qiu X, Du Y H, Lou B, et al. Synthesis and identification of new 4-arylidene curcumin analogues as potential anticancer agents targeting nuclear factor-κB signaling pathway[J]. J Med Chem, 2010, 53 (23): 8260-8273.
[10] Youssef D, Nichols C E, Cameron T S, et al. Design,synthesis, and cytostatic activity of novel cyclic curcumin analogues [J]. Bioorg. Med. Chem. Lett., 2007, 17(20):5624-5629.
[11] Fuchs J R, Pandit B, Bhasin D, et al. Structure-activity relationship studies of curcumin analogues [J]. Bioorg. Med. Chem. Lett., 2009, 19(7): 2065-2069.
[12] Zhou J, Geng G, Batist G, et al. Syntheses and potential anti-prostate cancer activities of ionone-based chalcones [J]. Bioorg. Med. Chem. Lett., 2009, 19(4): 1183-1186.
[13] Liang G, Shao L, Wang Y, et al. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents [J]. Bioorg Med Chem., 2009, 17(6): 2623-2631.
[14] Suarez J A Q, Rando D G, Santos R P, et al. New antitumoral agents I: In vitro anticancer activity and in vivo acute toxicity of synthetic 1, 5-bis (4-hydroxy-3-methoxyphenyl)-1, 4-pentadien-3-one and derivatives [J]. Bioorg Med Chem., 2010, 18(17): 6275-6281.
[15] Tan K L, Koh S B, Ee R P L, et al. Curcumin Analogues with Potent and Selective Anti-proliferative Activity on Acute Promyelocytic Leukemia: Involvement of Accumulated Misfolded Nuclear Receptor Co-repressor (N-CoR) Protein as a Basis for Selective Activity [J]. Chem Med Chem., 2012, 7(9): 1567-1579.
[16] Wei X, Du Z Y, Zheng X, et al. Synthesis and evaluation of curcumin-related compounds for anticancer activity [J]. Eur. J. Med. Chem., 2012(53): 235-245.
[17] Yadav B, Taurin S, Rosengren R J, et al. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin[J]. Bioorg Med Chem., 2010, 18(18): 6701-6707.
[18] Simoni E, Bergamini C, Fato R, et al. Polyamine conjugation of curcumin analogues toward the discovery of mitochondria-directed neuroprotective agents[J]. J Med Chem., 2010, 53(19): 7264-7268.
[19] Makarov M V, Leonova E S, Rybalkina E Y, et al. Synthesis,characterization and structure-activity relationship of novel N-phosphorylated E, E-3,5-bis(thienylidene) piperid-4-ones [J]. Eur J Med Chem., 2010(45): 992-1000.
[20] K á lai T, Kuppusamy M L, Balog M, et al. Synthesis of N-substituted 3, 5-bis (arylidene)-4-piperidones with high antitumor and antioxidant activity[J]. J Med Chem., 2011,54(15): 5414-5421.
[21] Das S, Das U, Michel D, et al. Novel 3, 5-bis(arylidene)-4-piperidone dimers: Potent cytotoxins against colon cancer cells [J]. Eur J Med Chem., 2013(64): 321-328.
[22] Das S, Das U, Sakagami H, et al. Dimeric 3,5-bis (benzylidene)-4-piperidones: A novel cluster of tumourselective cytotoxins possessing multidrug-resistant properties [J]. Eur J Med Chem., 2012(51): 193-199.
[23] Helal M, Das U, Bandy B, et al. Mitochondrial dysfunction contributes to the cytotoxicity of some 3, 5-bis (benzylidene)-4-piperidone derivatives in colon HCT-116 cells [J]. Bioorg Med Chem Lett., 2013, 23(4): 1075-1078.
[24] Zhang Q, Zhong Y, Yan L N, et al. Synthesis and preliminary evaluation of curcumin analogues as cytotoxic agents [J]. Bioorg Med Chem Lett., 2011, 21(3): 1010-1014. [25] Woo H B, Eom Y W, Park K S, et al. Synthesis of substituted benzimidazolyl curcumin mimics and their anticancer activity [J]. Bioorg Med Chem Lett., 2012, 22(2):933-936.
[26] Simoni D, Rizzi M, Rondanin R, et al. Antitumor effects of curcumin and structurally β-diketone modified analogs on multidrug resistant cancer cells [J]. Bioorg Med Chem Lett., 2008, 18(2): 845-849.
[27] Qiu P, Xu L, Gao L, et al. Exploring pyrimidine-substituted curcumin analogues: design, synthesis and effects on EGFR signaling [J]. Bioorg Med Chem., 2013, 21(7): 5012-5020.
Study Advances of Structural Modification and Antitumor Activities of Curcumin
LI Jia-qing
(Department of Biological Medicine, Changjiang Polytechnic, Wuhan 430064, China)
Curcumin was a kind of natural polyphenol widely found in natural plants and had various biological activities. However,anti-tumor effect was weak and bioavailability of curcumin was poor, which had limited the potential clinical application. Based on this fact, researchers had conducted a great deal of structural modifications on curcumin, in order to improve its anti-cancer activities and druggability. In this paper, the research advances of the structural modification of curcumin, as well as anti-cancer activities were reviewed, and those would provide assistances for the subsequent synthesis of curcumin derivatives with high and specific biological activity.
curcumin; structural modification; anti-cancer activity
R 282.71
A
1671-9905(2016)08-0030-06
李家庆(1963-),男,湖北武汉人,长江职业学院生物医药学院高级讲师,主要研究方向:新药开发和质量控制。
2016-06-15
