Preparation of Nitrogen-Doped Carbon Catalyst to Oxygen Reduction Reaction and Influence of Protective Gas Flowing on Its Activity
2016-09-13ZhongpingXiongYujunSiHongYuMinjiaoLiMaoxueChenKeyLaboratoryofGreenCatalysisofHigherEducationInstitutesofSichuanCollegeofChemistryandPharmaceuticalEngineeringSichuanUniversityofScienceandEngineeringZigong643000ChinaDa
Zhong-ping Xiong,Yu-jun Si,Hong Yu,Min-jiao Li,Mao-xue ChenKey Laboratory of Green Catalysis of Higher Education Institutes of Sichuan,College of Chemistry and Pharmaceutical Engineering,Sichuan University of Science and Engineering,Zigong 643000,China(Dated:Received on June 24,2015;Accepted on October 10,2015)
Preparation of Nitrogen-Doped Carbon Catalyst to Oxygen Reduction Reaction and Influence of Protective Gas Flowing on Its Activity
Zhong-ping Xiong,Yu-jun Si∗,Hong Yu,Min-jiao Li,Mao-xue Chen
Key Laboratory of Green Catalysis of Higher Education Institutes of Sichuan,College of Chemistry and Pharmaceutical Engineering,Sichuan University of Science and Engineering,Zigong 643000,China
(Dated:Received on June 24,2015;Accepted on October 10,2015)
A non-precious metal catalyst MnHMTA/C to oxygen reduction reaction was prepared by pyrolyzing a precursor from manganese chloride,hexamethylenetetramine and acetylene black in nitrogen gas atmosphere.The effect of heat treatment temperature and flowing of nitrogen gas were investigated.A catalyst with the highest activity can be obtained at 700◦C. Mn(II)ion was changed to MnO in heat treatment,which improved the catalytic activity of the catalyst.Hexamethylenetetramine takes part in the formation of active site of the catalyst as its decomposed gases.The flowing of protective gas takes the decomposed gases out of the tube furnace and brings negative effect on the catalytic activity of the MnHMTA/C catalyst.
Oxygen reduction reaction,Non-precious metal catalyst,Manganese,Protective gas flowing
I.INTRODUCTION
Platinum-containing catalysts are widely used to accelerate the hydrogen oxidation reaction(HOR)and oxygen reduction reaction(ORR)in fuel cells.Especially,the ORR is sluggish in kinetics and the reaction rate is slower than HOR at 6-7 order on the Pt catalyst.It always needs more Pt loading in the ORR catalyst to get greater current density on the cathode. However,the limited reserve and high cost of platinum block the large scale application of the fuel cells based on platinum catalyst[1-3].
In the past decades,considerable research efforts have been devoted to the development of non-precious metal and non-metal electrocatalysts for fuel cell applications[4-7].Among them,heat treated carbon based N4-macrocycle compounds,such as phthalocyanine and porphyrin with transition metal ions(such as Co(II),Fe(II)),have attracted intensive attentions.This type of catalyst is widely named as nitrogen-doped carbon catalyst[8-16].Now the catalyst can be prepared by pyrolyzing a solid precursor containing metal ions,carbon black and nitrogen compounds in an inert gas atmosphere.In the heat treating process,the nitrogen atoms would enter into the structure of carbon black to form new C-N structures,such as quaternary C-N,pyridinic C-N,pyrrolic C-N,and graphite C-N,some of which are viewed as the active sites of the catalyst[17-20].
It is comprehensible that most nitrogen-containing organic compounds would be decomposed at high temperature in heat treatment.The decomposed products may include some small gas molecules containing nitrogen atom.If the active sites of catalyst were formed in the reaction between gas molecules and carbon black,the flowing of the gases would influence the formation of the active sites.Additionally,the flowing of the reactive gases can be influenced by the flowing of inert gas which usually acts as protective gas to avoid the oxidation of carbon black.So the flowing of protective gas would influence the catalytic activity of the resulted catalyst.In this work,a nitrogen-doped ORR catalyst was prepared by heat treating a precursor from manganese chloride(MnCl2),hexamethylenetetramine(HMTA)and acetylene black in nitrogen gas atmosphere by an improved method proposed in our previous work[21,22].The influence of protective gas flowing was also investigated.
II.EXPERIMENTS
A.Preparation of catalyst
The catalyst was synthesized by pyrolyzing a precursor from MnCl2·4H2O,hexamethylenetetramine and acetylene black with a mass ratio of 1∶2∶2.For the preparation of the precursor,MnCl2was dissolved in ethanol.Then acetylene black was mixed into the solution to obtain a paste.The paste was pre-dried in air before moving into an agate mortar and dried at 100◦C for 2 h.By this method,the Mn(II)ions can be uniformly dispersed on the surface of acetylene black.Then hexamethylenetetramine was added into the agate mortar and ground for 30 min to get a precursor.The precursor was treated in tube furnace using nitrogen as protective gas.The nitrogen gas was controlled by two modes in the heat treatment.One mode is that the nitrogen gas keeps flowing through the tube furnace in the whole heat treatment.The other is that the nitrogen gas is closed after heat treating temperature reached a specific value.In the heat treatment,the temperature was ramped at a rate of 10◦C/min and kept for 2 h at a set value.Then the tube furnace was naturally cooled. The resulted catalyst was labeled as CoHMTA/C.

FIG.1 The SEM imagines of acetylene black and MnHMTA/C catalysts obtained at different heat treating temperatures.(a)Acetylene black,(b)500◦C,(c)600◦C,(d)700◦C,and(e,f)800◦C.
B.Characterization of catalyst and evaluation on activity
The morphologies of the samples were characterized using scanning electron microscope imagines(SEM)collected on a TESCAN VEGA 3 at 20 kV.The phase of manganese in the catalyst was characterized by X-ray diffraction(XRD).XRD patterns were recorded on a DX-2600 X-ray diffractometer using Cu Kα(λ=0.15406 nm)radiation and equipped with a graphite monochromator at a scanning rate of 4◦/min. The X-ray tube was operated at 40 kV and 25 mA. The catalytic activity of the catalyst was evaluated by ORR onset potential and limit current from linear sweep voltammetry(LSV)tested in 0.5 mol/L H2SO4solution saturated by oxygen.The electrochemical test was carried out on a CHI760E workstation with a saturated calomel electrode as the reference electrode,a platinum sheet as the counter electrode.The working electrode was fabricated as follows∶10 mg of the catalyst,1.0 mL of isopropyl alcohol,1.0 mL of deionized water,and 0.1 mL of 0.5wt%Nafion solutions were mixed and ultrasonicated for 20 min to form a uniform ink.Then,10µL of the ink was added on the clean surface of a glassy carbon disk(Φ 5 mm).The coated electrode was naturally dried for 2 h before test.The LSV test was conducted at a scan rate of 5 mV/s at 30◦C.
III.RESULTS AND DISCUSSION
A precursor from MnCl2·4H2O,hexamethylenetetramine and acetylene black was heat treated at 500,600,700,800,and 900◦C for 2 h to investigate the influence of temperature on the catalytic performance. The protective nitrogen gas kept flowing in the whole process.Figure 1 shows the SEM images of untreated acetylene black and MnHMTA/C catalysts obtained at 500,600,700,and 800◦C.It can be seen the particle size of acetylene black is about 50-100 nm and there are some bigger aggregates about 200 nm in the untreated acetylene black.These aggregates gradually disappeared after the high temperature heat treating. But other obvious difference between the five samples can not be found,even in a greater magnified SEM(Fig.1(f)).That is to say that the influences of HMTA and MnCl2can not be detected by SEM.The reason is that the HMTA would decompose to gases and the gases react with acetylene black to form the C-N structures.The reaction does not change the morphology of the acetylene black.And in the preparation of the MnHMTA/C catalyst,MnCl2was dissolved in ethanoland then was highly dispersed on the surface of acetylene black,so it is also not be viewed in the SEM images.
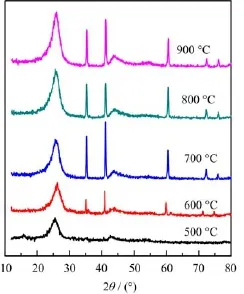
FIG.2 The XRD patterns of MnHMTA/C catalysts obtained at different heat treating temperatures.

FIG.3The LSV curves of MnHMTA/C catalysts obtained at different heat treating temperatures.(a)500◦C,(b)600◦C,(c)700◦C,(d)800◦C,and(e)900◦C.
The XRD patterns of the catalysts obtained at different temperatures are shown in Fig.2.The wide diffraction peak at 25.6◦can be attributed to acetylene black and the other narrow peaks at 35.0◦,40.7◦,58.9◦,70.4◦,and 74.0◦belong to the diffraction peaks of MnO(PDF No.07-0230).According to our previous studies,the Co(II)ions can be reduced to metallic state by carbon black at a high temperature in an inert atmosphere[21]. But in this work,the obvious diffraction peaks of metallic manganese can not be indexed in the MnHMTA/C catalysts,which shows that it is difficult to reduce the Mn(II)ions,or the metallic manganese are prone to be oxidized by the trace oxygen in the protective gas.
Figure 3 shows the LSV curves of the catalysts,respectively.The ORR onset potential and limit current can be used to evaluate the catalytic activity of the catalyst.More positive ORR onset potential and larger limit current reflect better catalytic activity.The heat treating temperature has obvious influence on the performance of the MnHMTA/C catalyst.The catalyst obtained at 500◦C has little activity to ORR.The activity of catalyst obtained at 600◦C shows an obvious improvement with an onset potential about 0.47 V(vs. SCE).A best catalyst was prepared at 700◦C with an onset potential about 0.57 V(vs.SCE)and larger limit current.By further elevating the temperature to 800 and 900◦C,the ORR onset potential does not change,but the limit current gradually decreases.
It can be observed that there exists a correlation between MnO and the ORR catalytic activity(Fig.2 and Fig.3).The diffraction peaks of MnO can not be found in the XRD pattern of catalyst obtained at 500◦C and the catalytic activity is worse.In the XRD pattern of catalyst obtained at 600◦C,the diffraction peaks of MnO appears and the catalyst exhibits obvious activity.With the increase of diffraction peak intensities in the catalyst obtained at 700◦C,the catalyst shows better activity.However,the diffraction peaks keep strong in the catalysts obtained at 800 and 900◦C,but both of their catalytic activities are worse than the catalyst obtained at 700◦C.These results indicate that there is a relation between MnO and the formation of active site of MnHMTA/C catalyst,but MnO itself may not be the active site of the catalyst.
In order to investigate the function of MnO further,the catalyst obtained at 700◦C was immersed in 0.5 mol/L H2SO4solution for 1 h.Figure 4 shows the XRD patterns and LSV curves of the catalyst before and after the immersion.It can be seen that MnO in the catalyst is leached out after the immersion,but the catalytic activity of the catalyst doesn’t deteriorate because of the leaching out of MnO.The result further conforms MnO is not the active site of the catalyst.
In this work,nitrogen-containing compound hexamethylenetetramine was used as nitrogen source in the preparation of the MnHMTA/C catalyst.According to the physical properties of hexamethylenetetramine,it would sublimate to gas at 263◦C and would decompose and release HCN gas at 300◦C.Other gases would be released at higher temperature.So it can be speculated that hexamethylenetetramine would participate in the formation of the active site of the catalyst as its decomposed product.In order to conform this idea,several catalysts were prepared by pyrolyzing the precursor at 700◦C for 2 h.In heat treating process,the protective nitrogen gas was closed when the temperature was elevated to 300,400,500,600,and 700◦C,respectively. The LSV curves of the resulted catalysts are shown in Fig.5.It can be seen that the ORR onset potentials of different catalysts are equal to each other,but the limit currents are different.The limit current is larger when the nitrogen gas was closed at lower temperature.
However,the limit current decreases by closing protective gas at higher temperature or not closing.Theresult shows the speculation that the hexamethylenetetramine participates in the formation process of the active site of the catalyst as its decomposed product is true.If the protective gas was closed at a lower temperature,the decomposed gases of hexamethylenetetramine would retain in the tube furnace with a larger concentration.The larger concentration of reactants ensures that more active sites are formed on the surface of catalyst in the same duration.In contraries,the protective gas was closed at a higher temperature or not be closed,part of decomposed gases of hexamethylenetetramine would be taken out of the tube furnace with the flowing of protective gas,which reduced the effective concentration of gaseous reactants in the formation process of active site.So the less active site would be formed on the surface of catalyst and the activity of the resulted catalyst would also be deteriorated.

FIG.4(a)XRD patterns of catalyst obtained at 700◦C before and after acid immersion in 0.5 mol/L H2SO4solution for 1 h.(b)LSV curves of catalyst obtained at 700◦C before and after acid immersion 0.5 mol/L H2SO4solution for 1 h.

FIG.5 The LSV curves of the catalysts prepared by closing protective nitrogen gas at different temperature.
IV.CONCLUSION
A non-precious metal catalyst MnHMTA/C to ORR can be prepared by heat treating a precursor from manganese chloride,hexamethylenetetramine,and acetylene black.The hexamethylenetetramine decomposes and releases nitrogen-containing gases during the pyrolysis.These gases can react with acetylene black to form the active site of the catalyst.The Mn(II)ion was changed to MnO in the heat treating process.There is a close correlation between MnO and the catalytic activity of the MnHMTA/C catalyst,but the MnO itself is not a part of the active site of the catalyst.It is important to control the flowing of protective gas during the heat treatment.The flowing protective gas takes some of the decomposed gases of hexamethylenetetramine out of tube furnace and decreases the effective concentration of gases in the tube furnace.This effect induces less active site which was formed on the surface of the catalyst and the catalyst shows worse catalytic activity to ORR.
V.ACKNOWLEDGEMENTS
This work is supported by the Specialized Research Fund for the Doctoral Program of Sichuan University of Science and Engineering(No.2012RC16),the Opening Project of Key Laboratory of Green Catalysis of Sichuan Institutes of High Education(No.LYJ1401 and No.LYJ14206),and the Program of Education Department of Sichuan Province(No.15ZB0208).
[1]S.C.Thomas,X.Ren,S.Gottesfeld,and P.Zelenay,Electrochim.Acta 47,3741(2002).
[2]A.Serov and C.Kwak,Appl.Catal B 90,313(2009).
[3]V.Nallathambi,J.W.Lee,S.P.Kumaraguru,G.Wu,and B.N.Popov,J.Power Sources 183,34(2008).
[4]B.Wang,J.Power Sources 152,1(2005).
[5]M.Lefevre,E.Proietti,F.Jaouen,and J.P.Dodelet,Science 324,71(2009).
[6]D.Z.Mezalira and M.Bron,J.Power Sources 231,113(2013).
[7]F.Jaouen,E.Proietti,M.Lef`evre,R.Chenitz,J.P. Dodelet,G.Wu,H.T.Chung,C.M.Johnston,and P. Zelenay,Energ.Environ.Sci.4,114(2011).
[8]H.Li,W.Kang,L.Wang,Q.Yue,S.Xu,H.Wang,and J.Liu,Carbon 54,249(2013).
[9]H.R.Byon,J.Suntivich,and Y.Shao-Horn,Chem. Mater.23,3421(2011).
[10]R.F.Wang,T.B.Zhou,H.Li,H.Wang,H.Q.Feng,J.Goh,and S.Ji,J.Power Sources 261,238(2014).
[11]K.Gong,F.Du,Z.H.Xia,M.Durstock,and L.Dai,Science 323,760(2009).
[12]G.Wu,K.L.More,C.M.Johnston,and P.Zelenay,Science 332,443(2011).
[13]I.Kruusenberg,L.Matisen,Q.Shah,A.M.Kannan,and K.Tammeveski,Int.J.Hydrogen Energy 37,4406(2012).
[14]I.Kruusenberg,L.Matisen,and K.Tammeveski,J. Nanosci Nanotechnol.13,621(2013).
[15]I.Kruusenberg,S.Ratso,M.Vikkisk,P.Kanninen,T. Kallio,A.M.Kannan,and K.Tammeveski,J.Power Sources 281,94(2015)
[16]F.He,J.Yang,R.Li,B.H.Liu,and Z.P.Li,J.Power Sources 274,48(2015).
[17]P.H.Matter,E.Wang,M.Arias,E.J.Biddinger,and U.S.Ozkan,J.Mol.Catal.A 264,73(2007).
[18]K.Artyushkova,S.Levendosky,P.Atanassov,and J. Fulghum,Top Catal.46,263(2007).
[19]H.Niwa,M.Saito,M.Kobayashi,Y.Harada,M. Oshima,S.Moriya,K.Matsubayashi,Y.Nabae,S. Kuroki,T.Ikeda,K.Terakura,J.Ozaki,and S.Miyata,J.Power Sources 223,30(2013).
[20]Y.Hu,X.Zhao,Y.Huang,Q.Li,N.J.Bjerrum,C. Liu,and W.Xing,J.Power Sources 225,129(2013).
[21]Y.J.Si,Z.P.Xiong,C.G.Chen,P.Liu,and H.J.Wu,Chin.Chem.Lett.24,1109(2013).
[22]Y.J.Si,C.G.Chen,W.Yin,and H.Cai,Chin.Sci. Bull.56,1086(2011).
∗Author to whom correspondence should be addressed.E-mail:syj08448@163.com
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Raman Spectra of Liquid Nitromethane under Singly Shocked Conditions
- Tunneling Electron Induced Fluorescence from Single Porphyrin Molecules Decoupled by Striped-Phase Octanethiol Self-assembled Monolayer
- Ion Product of Pure Water Characterized by Physics-Based Water Model
- Laser Linewidth and Spectral Resolution in Infrared Scanning Sum Frequency Generation Vibrational Spectroscopy System
- Performances of Five Representative Force Fields on Gaseous Amino Acids with Different Termini
- Dynamics of Tripartite Entanglement and Intramolecular Energy in Symmetric Trimer Molecule