Performances of Five Representative Force Fields on Gaseous Amino Acids with Different Termini
2016-09-13XinChenZijingLinHefeiNtionlLbortoryforPhysiclSciencestheMicrosclendCASKeyLbortoryofStronglyCoupledQuntumMtterPhysicsUniversityofSciencendTechnologyofChinHefei230026ChinKeyLbortoryofMterilsPhysicsInstituteofSolidStt
Xin Chen,Zi-jing Lin,b∗.Hefei Ntionl Lbortory for Physicl Sciences t the Microscle nd CAS Key Lbortory of Strongly-Coupled Quntum Mtter Physics,University of Science nd Technology of Chin,Hefei 230026,Chin b.Key Lbortory of Mterils Physics,Institute of Solid Stte Physics,Chinese Acdemy of Sciences,Hefei 230031,Chin(Dted:Received on July 15,2015;Accepted on September 8,2015)
Performances of Five Representative Force Fields on Gaseous Amino Acids with Different Termini
Xin Chena,Zi-jing Lina,b∗
a.Hefei National Laboratory for Physical Sciences at the Microscale and CAS Key Laboratory of Strongly-Coupled Quantum Matter Physics,University of Science and Technology of China,Hefei 230026,China b.Key Laboratory of Materials Physics,Institute of Solid State Physics,Chinese Academy of Sciences,Hefei 230031,China
(Dated:Received on July 15,2015;Accepted on September 8,2015)
There is a growing interest in the study of structures and properties of biomolecules in gas phase.Applications of force fields are highly desirable for the computational efficiency of the gas phase study.To help the selection of force fields,the performances of five representative force fields for gaseous neutral,protonated,deprotonated and capped amino acids are systematically examined and compared.The tested properties include relative conformational energies,energy differences between cis and trans structures,the number and strength of predicted hydrogen bonds,and the quality of the optimized structures.The results of BHandHLYP/6-311++G(d,p)are used as the references.GROMOS53A6 and ENCADS are found to perform poorly for gaseous biomolecules,while the performance of AMBER99SB,CHARMM27 and OPLSAA/L are comparable when applicable.Considering the general availability of the force field parameters,CHARMM27 is the most recommended,followed by OPLSAA/L,for the study of biomolecules in gas phase.
Conformation,Relative energies,Correlation coefficient,Hydrogen bond,Molecular mechanics
I.INTRODUCTION
Empirical molecular force fields are routinely used in the investigations of structure-property-activity relationships in biological systems as the computational cost of treating these systems quantum mechanically is often unbearably high.The force fields are in general derived by fitting parameters to data from quantum chemical calculations or experiments on model molecules that may mimic the properties of the interested biomolecules.Fitting the experimental data for the condensed-phase environment is emphasized. This is reasonable as the structures and properties of biomolecules in solution or condensed-phase environment are of the most interest.However,this also means that the accuracy of the force field to predict the relevant properties in the gas phase is sacrificed.It also represents a paradox in the fitting philosophy as the quantum chemistry results for the gas phase are indispensible for the force field parameterization[1].
The limitation of force fields for the gas phase study is often dismissed as irrelevant.Indeed,impressive progresses have been made in utilizing force fields for the study of biological systems,e.g.,simulating the protein dynamics[2-7].However,the contradiction inherent in the fitting philosophy has some serious consequences. As there is no rigorous way to determine the optimal parameter set,many force field variants are proposed based on different emphases of the fitting targets.AMBER[8],CHARMM[9],ENCAD[10],GROMOS[11]and OPLSAA[12]are examples of force fields reports that are widely in use.There have been numerous articles and reviews discussing about the performances of modern force fields for biomolecules in solution or condensed-phase environment[4,5,7,8,13].These studies show clearly that a proper choice of the force field is dependent on the research subject of interest.
Comparative studies of the force fields are crucial for the proper selection of force field,but systematic studies on the performance comparison for objects in gas phase are rare.The limitation of force fields for the gas phase study had been dismissed as irrelevant.However,there is a growing interest and effort in studying the gaseous biomolecules that are free from the complication caused by the complex solute-solvent interactions[14-20].Consequently,a systematic comparison of the performances of the force fields for biomolecules in gas phase is becoming increasingly meaningful.The comparison is helpful for the choice of force field that is necessary for many gas phase studies,e.g.,MD runs of biomolecule with about 100 or more atoms.Evenwhen the force field is used as a pre-screening tool,the comparative study is also helpful for avoiding using a force field that provides misguided information about the potential energy surface.Moreover,the comparative study is the starting point for learning the accuracies of the state-of-the-art force fields for the gas phase study.Such information is also helpful for knowing how much the improvement of force field is required to provide results for the gas phase study with a desirable accuracy.The information is useful for guiding the future development of force field.
The choice of testing objects in this study is guided by some general considerations.First,protein force field parameters are mainly determined by fitting data for capped alanine,capped glycine and capped proline[13,14,21-24].Other amino acid residues play only minor roles in the parameter determination.The approach seems to work well for proteins in condensedphase environment[2,5,7,14,18],partly due to the fact that most attention is paid to the backbone structures.For short peptides,the influence of the side chain on the backbone is expected to be substantial and how well the force fields work for other amino acid residues requires detailed examinations.Secondly,the overall influence of the amino and carboxyl terminal groups is relatively small for large molecules such as proteins,but the corresponding influence for short peptides can be significant.Short peptides are a class of biomolecules with important biological and physiological roles.They are also the starting point for the peptide-based drug design study[25].The amino and carboxyl groups are important in the force field performance study.Thirdly,proton transfer is critically important for numerous biological processes.It is therefore highly meaningful to include the protonated amino group and deprotonated carboxyl group in the testing object set.Therefore,the force field performances are tested here for a large number of gaseous amino acids with natural,protonated,deprotonated and capped termini.
Force fields are meant to reproduce the structural and energetic information obtained by experiments,preferably,or quantum chemistry calculations.As the experimental data are very limited,comparing the force fields with the quantum chemistry calculations is more convenient and,possibly,statistically more meaningful. In fact,the best one may hope for that the results by a force field are close to that by quantum chemistry calculations.Therefore,the quantum chemistry results may be used as the references to test the performances of different force fields.Notice that there are a number of quantum chemistry methods and the density functional theory(DFT)based approach is favored for its accuracy and computational efficiency.Among the DFT variants tested for all neutral,protonated and deprotonated amino acids,the BHandHLYP/6-311++G(d,p)method has been shown to produce the best energetic results statistically[26].When benchmarked with the CCSD/6-311++G(d,p)results,the statistical quality of the BHandHLYP results is even slightly better than that of the MP2 computations[26].Consequently,the force fields are benchmarked with the BHandHLYP results.
II.METHOD
The abilities of five force fields,AMBER,CHARMM,ENCAD,GROMOS and OPLSAA,to mimic the quantum chemistry,BHandHLYP/6-311++G(d,p)energetic and geometric results are tested with four representative properties∶(i)relative conformational energies,(ii)energy differences between cis and trans structures,(iii)number of predicted hydrogen bonds(H-bonds),(iv)structural similarity as measured by the root-meansquare-difference(RMSD).The five force fields have various versions of parameterizations and their relatively new parameterization sets,AMBER99SB[27],CHARMM27[21],GROMOS53A6[28],ENCADS[29],OPLSAA/L[30],are used in the test.
Nineteen naturally occurring amino acids including glycine(Gly or G),alanine(Ala or A),valine(Val or V),leucine(Leu or L),isoleucine(Ile or I),asparagine(Asn or N),glutamine(Gln or Q),cysteine(Cys or C),methionine(Met or M),serine(Ser or S),threonine(Thr or T),proline(Pro or P),tyrosine(Tyr or Y),tryptophan(Trp or W),phenylalanine(Phe or F),histidine(His or H),lysine(Lys or K),aspartic acid(Asp or D)and glutamic acid(Glu or E)are considered in the testing set of the current study.Four terminus forms of amino acids,natural,protonated,deprotonated and capped,are used in the testing.In capped amino acids,the N-and C-termini are capped with the acetyl and N-methylamine groups,respectively.To illustrate,the structures of natural,protonated,deprotonated and capped alanine are sketched in Fig.1.
To be of high statistical significance,the conformations of these amino acids are obtained through systematic searches by considering all combinations of bond rotational degrees of freedom in the trial structure generations,as described in Ref.[31].The quantum mechanics(QM)structures are determined at the BHandHLYP/6-31G∗level.The numbers of unique conformations thus obtained for these molecules are shown in parentheses as the followings.For natural,protonated,deprotonated and capped amino acids,they are respectively Ala(8,3,2,6),Asn(62,9,11,71),Asp(106,26,24,20),Cys(78,5,12,49),Gln(59,8,14,138),Glu(340,10,16,72),Gly(16,3,1,4),His(57,12,8,56),Ile(85,33,19,62),Leu(96,12,23,68),Lys(186,13,51,427),Met(172,15,31,190),Phe(31,10,8,32),Pro(20,6,7,10),Ser(52,12,19,49),Thr(71,12,7,52),Trp(67,5,16,60),Tyr(73,10,36,58),Val(37,14,10,19).
The quality of relative conformational energies is measured with three quantities,the average absolute error(AAE),the maximum absolute error(MAE),and the correlation coefficient(CC).AAE and MAE for anamino acid are defined as∶

The correlation coefficient,CC,for an amino acid is calculated as[32],
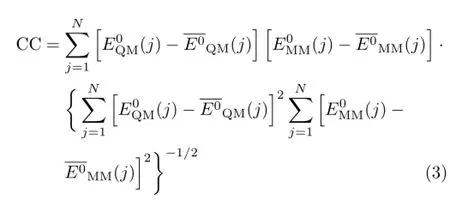
The energy difference between cis and trans structures is tested on three amino acids,Ala,Asn and Cys,that are representative of amino acids with aliphatic,amido and sulfur side chains.The trial structures were generated by considering all combinations of their bond rotational degrees of freedom.If the potential energy surfaces determined by QM and MM are similar,the MM relative conformational energy is expected to have a linear relationship with the QM relative conformational energy.Consequently,the cis-trans energy difference is tested by using the linear fit with the least square difference[33],

Here X(Y)is the QM(MM)value of the cis-to transcarboxylic relative energy,a and b are coefficients of the linear fitting.The quality of the linear fit is measured by the coefficient of determination(COD)[34]R,
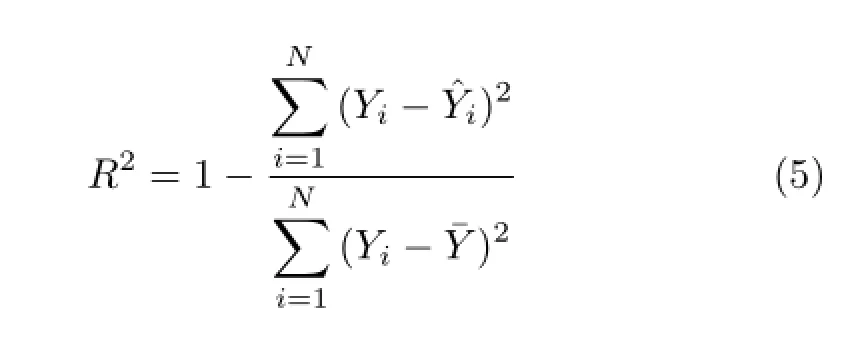
Here Yi(ˆYi)is the QM(MM linear fit)result for the cis-to trans-carboxylic relative energy of configuration i and¯Y is the average of Yifor the considered configurations.When R is acceptably large(close to 1),the linear regression coefficients of QM and force fields,a’s,are compared to determine the performances of the force fields for describing the relative energies of cis and trans-carboxylic configurations.

FIG.1 A sketch of alanine with different terminus.(a)Neutral alanine,(b)protonated alanine,(c)deprotonated alanine,(d)capped alanine.
The number of H-bonds is counted over all conformations of all amino acids of a specific terminus type.An H-bond is found if the distance between a donor and an acceptor is less than 2.8˚A.For natural amino acids,the H-bonds are counted only between NH-OT and N-HO(Fig.1).For protonated amino acids,only the NH-OT H-bonds are counted(Fig.1(b)).For deprotonated and capped amino acids,the H-bond is formed between CO-NH pair,as shown in Fig.1(c)and(d),respectively.The number of H-bonds in amino acid conformations for a force field is counted with all the structures obtained by reoptimizing the QM conformations by the force field.The average length of the H-bonds is also calculated so that the average strength of the predicted H-bonds may be estimated.
In addition to comparison the H-bonds in the QM and MM structures,the RMSD of the QM and MMstructures is also examined.The RMDS between a QM and MM structure pair is defined as,
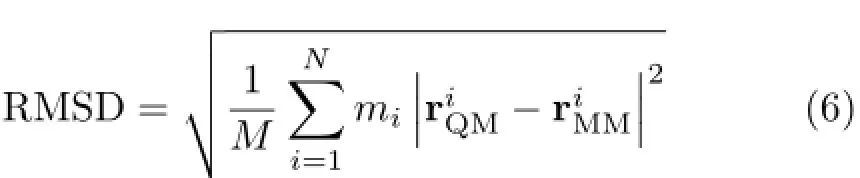
The BHandHLYP calculations were performed with the Gaussian 09 suite of programs[22].All other calculations were carried out with the Gromacs 4.5 program[35,36].

TABLE I AAE and MAE analysis for AMBER99SB(A99SB),CHARMM27(C27),ENCADS,GROMOS53A6(G53A6),OPLSAA/L(OL)on the relative conformational energies of 19 amino acids using the BHandHLYP/6-311++G(d,p)results as the references(in kcal/mol).
III.RESULTS AND DISCUSSION
A.Relative conformational energies
Table I summarizes the AAE and MAE results for the five force field parameter sets on the relative energies of all conformations of 19 amino acids.As seen in Table I,MAE and AAE are often closely correlated,i.e.,a high(low)MAE often means a high(low)AAE and vice versa.However,MAE seems to be more reflective about the extent of error that may be caused by using a force field,while such information may be buried in the AAE result due to the conformational averaging.In fact,all AAE values in Table I seem to be relatively small and acceptable.However,the MAE results show that very large error may be encountered for the conformations of some amino acids.The large difference between the AAE and MAE values indicates that the force fields may do well for most amino acids,but may perform very poorly for some amino acids.Overall,Table I shows that the performances of OPLSAA/L,AMBER99SB and CHARMM27 are clearly better than that of ENCADS and GROMOS53A6.Moreover,the performances of OPLSAA/L,AMBER99SB and CHARMM27 for neutral amino acids are better than that for capped amino acids.This is somewhat unexpected as that the capped amino acids are used as the model system for deducing the parameter set.One reason for the result may be that the parameter sets are designed to reproduce the condensed phase properties.Another reason may be that the poor results on capped amino acids are caused by the poor parameterization for lysine,as clearly indicated in Table I.In other words,improving the parameters for lysine in the parameter sets of OPLSAA/L,AMBER99SB and CHARMM27 is highly desirable.Besides,it is interesting to note that the parameter sets of OPLSAA/L,AMBER99SB and CHARMM27,though aimed at the condensed phase properties,seem to work reasonably well for the relative conformational energies.
To reveal more details about the force field performances,Fig.2 shows the AAEs of the five force fields for each individual amino acid.As shown in Fig.2,the poor performance of GROMOS53A6 is mainly caused by two amino acids.If the parameter sets for valine and leucine,and possibly threonine,are properly revised,the performance of GROMOS53A6 will be significantly improved.Though not as bad as GROMOS53A6 for valine and leucine,the performance of ENCADS is poor for quite a few amino acids.The biggest gain may be achieved by improving the ENCADS parameters for threonine,serine,proline,aspartic acid and lysine.
As seen in Table I as well as in Fig.2,the performances of OPLSAA/L and CHARMM27 may be most benefitted from improving parameters for deprotonated threonine,protonated aspartic acid and neutral proline. For AMBER99SB,the parameters for neutral valine should be improved.It is worth repeating that improving the parameters for capped lysine is important for all AMBER99SB,CHARMM27 and OPLSAA/L force fields.
While AAE and MAE may provide information about the overall error of estimating relative conforma-tional energies,the correlation coefficient can reveal the strength and direction of the linear relationship between the QM and MM results[28].Figure 3 shows the CC values of amino acids in four terminus types.Only CCs that pass the significance test and have values above 0.6 are reported in the figure.
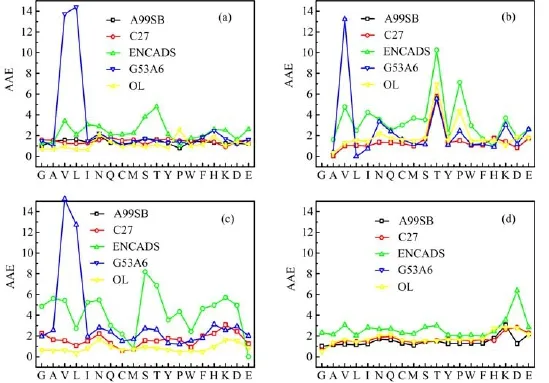
FIG.2 AAEs of AMBER99SB(A99SB),CHARMM27(C27),ENCADS,GROMOS53A6(G53A6)and OPLSAA/L(OL)for(a)neutral amino acids,(b)deprotonated amino acids,(c)protonated amino acids,and(d)capped amino acids.
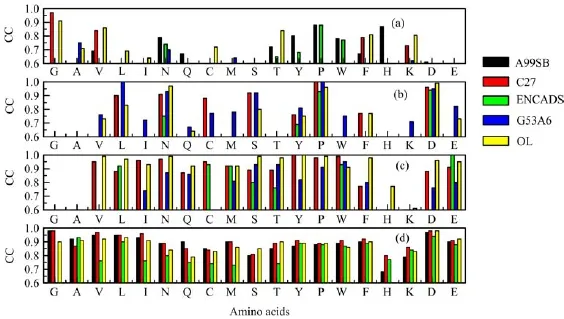
FIG.3 Correlation coefficients,CCs,of the AMBER99SB(A99SB),CHARMM27(C27),ENCADS,GROMOS53A6(G53A6)and OPLSAA/L(OL)force fields for the relative conformational energies of(a)neutral amino acids,(b)deprotonated amino acids,(c)protonated amino acids,and(d)capped amino acids.
Unlike the results shown in Table I,the best performing molecular type for the force fields as measured by CC is the capped amino acids,followed by the protonated amino acids,while the neutral amino acids are the worst performing molecules.The results are consistent with the fact that capped amino acids are used in the training set to deduce the force field parameters.It also shows that,although the overall error in the relative conformational energies is reasonably small,the orderings of the MM conformational energies for neu-tral amino acids are quite different from that of the QM results.
Combining Table I,Fig.2,and Fig.3,it may be said that all AMBER99SB,CHARMM27 and OPLSAA/L work very well for capped amino acids in gas phase. For protonated amino acids,OPLSAA/L works the best,followed by CHARMM27,then by AMBER99SB. OPLSAA/L also works well for neutral Gly,Ala,Val,Ile,Cys,Thr,Phe and Lys.For neutral Asn,Tyr,Pro,Trp and His,however,AMBER99SB performs the best.
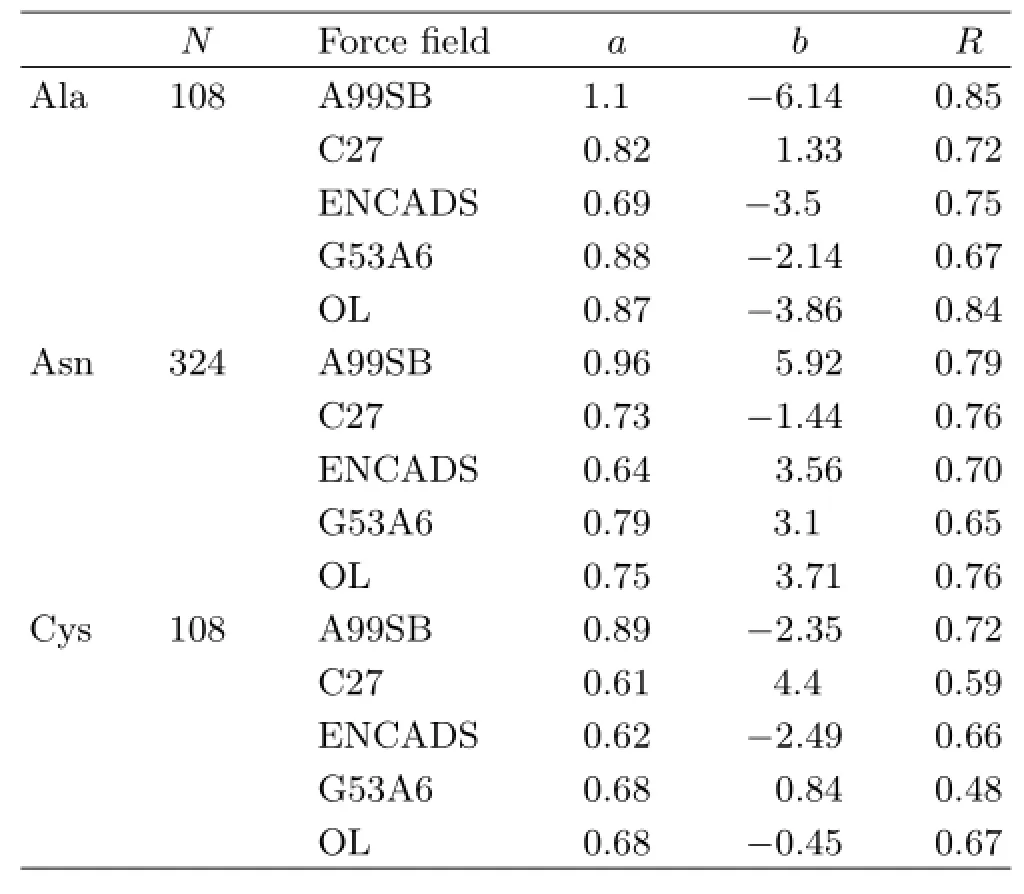
TABLE II Statistics of linear fitting between QM and MM results for the energy difference(in kcal/mol)between cisand trans-carboxylic configurations.
B.Energy difference between cis-and trans-carboxyl configurations
Table II shows the force field results on the relative energy between cis and trans carboxyl configurations for neutral Ala,Asn and Ser.Due to the large number of conformations used in the test,it is unrealistic to expect a high quality linear fit of the QM and MM results. Indeed,only modest CODs,R,are found in Table II. The average R for AMBER99SB,CHARMM27,ENCADS,GROMOS53A6 and OPLSAA/L is 0.79,0.69,0.71,0.60,and 0.76,respectively.Using a cutoff value of 2/3 for the average R,the linear fitting of GROMOS53A6 with QM may be rejected.
The linear fitting coefficient a is an indicator for the similarity between the QM and MM energy difference. The closer a is to one,the higher the similarity is. The average absolute deviation of a from one is 0.083,0.28,0.35 and 0.23 for AMBER99SB,CHARMM27,ENCADS and OPLSAA/L,respectively.The deviation for ENCADS is larger than 1/3.Overall,it may be said that the cis-trans energy difference is described the best by AMBER99SB,followed by OPLSAA/L,then by CHARMM27.The results by ENCADS and GROMOS53A6 are not very meaningful.The results for the cis-trans energy difference test are not identical to,but correlate reasonably with the results for the relative conformational energy difference test discussed above.
C.Hydrogen bonds
Here we focus on discussing the hydrogen bonds in the main chain of amino acids.The number counts and the average bond lengths of the H-bonds defined above as determined by QM and MM are summarized and compared in Table III.As the number of conformations considered is quite big and the conformations are about different amino acids,the results should be statistically meaningful.
As seen in Table III,all AMBER99SB,CHARMM27 and OPLSAA are more inclined to form the N-HO H-bond in neutral amino acids than that of the QM method.As indicated by the average H-bond length,OPLSAA/L and AMBER99SB tend to overestimate the N-HO H-bond strength,while the opposite is true for CHARMM27.OPLSAA/L is also more inclined to form the OT-HN H-bond than the QM result,but their H-bond strength is similar.CHARMM27,however,disfavors the OT-HN H-bond formation.The OT-HN H-bond strength predicted by CHARMM27 is also relatively low.AMER99SB seems to be the best for describing the OT-HN H-bond,though the bond strength is slightly underestimated.
AMBER99SB,CHARMM27,OPLSAA/L all describe well the CO···HN H-bond of capped amino acid. The best description is provided by CHARMM27,followed by OPLSAA/L.AMBER99SB tends to overestimate the CO···HN H-bond strength and the bond formation tendency.
Fordeprotonatedaminoacids,thenumberof the CO-HN H-bonds predicted by CHARMM27 is very similar to that of the QM result.However,CHARMM27 underestimates the H-bond strength. OPLSAA/L,on the other hand,vastly overestimates the tendency of forming the CO-HN H-bond,but gives a good description of the H-bond strength.
For protonated amino acids,both CHARMM27 and OPLSAA/L underestimate the OT-HN H-bond strength.However,CHARMM27 provides a good description for the H-bond formation tendency,while OPLSAA/L clearly overestimates the H-bond formation tendency.
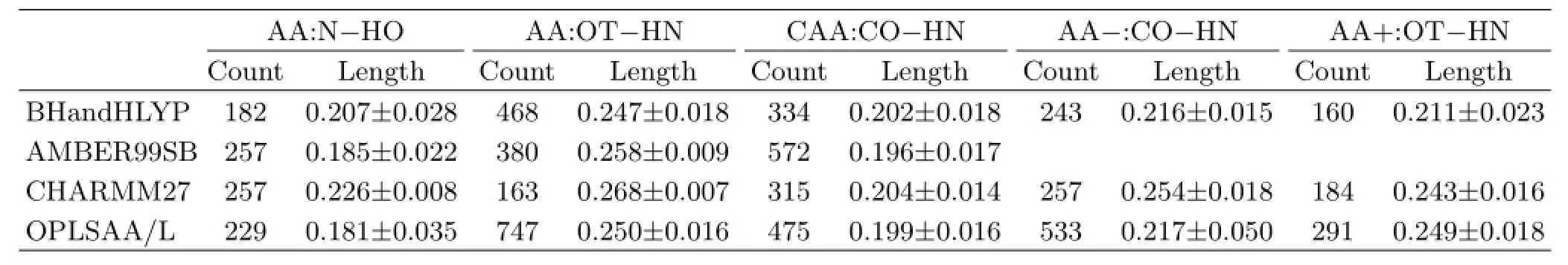
TABLE III Number count and average bond length(in nm)of the H-bonds predicted by QM and MM methods.
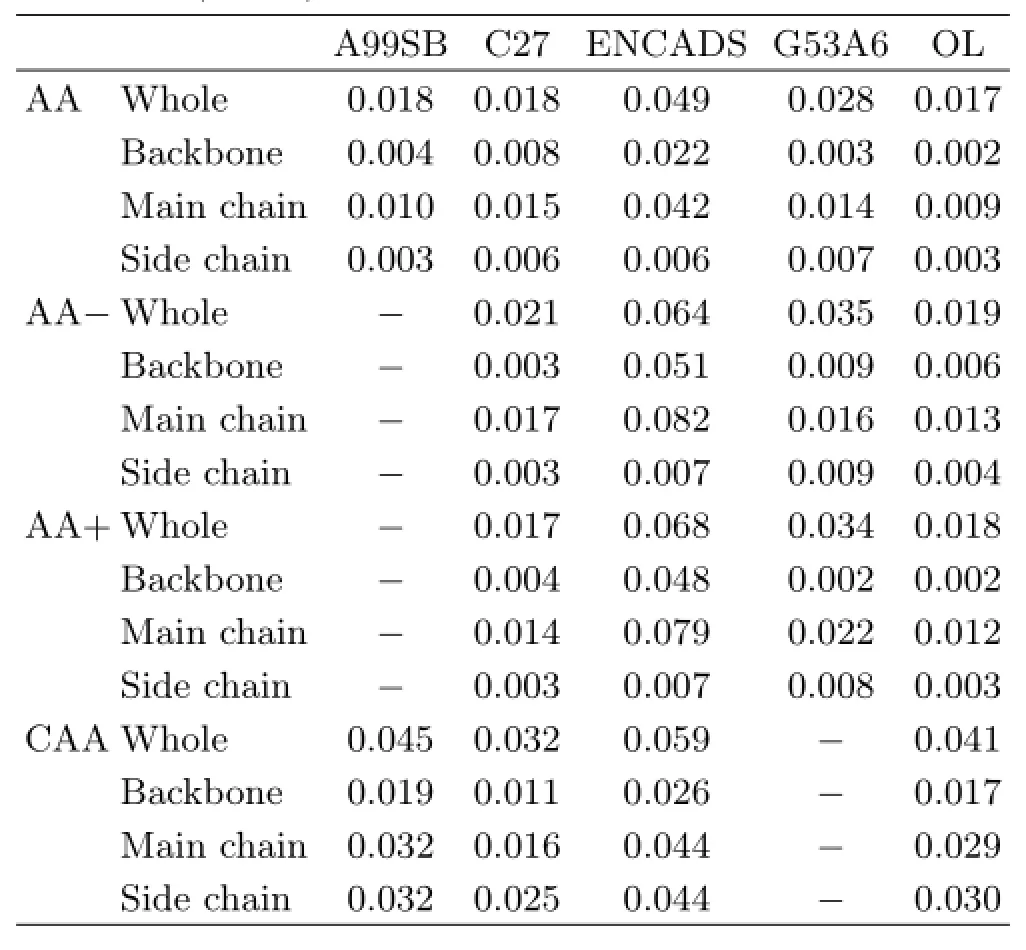
TABLE IV RMSD analysis on the MM structures when using the BHandHLYP/6-311++G∗∗conformations as the references(in nm).
D.RMSD
To examine the changes made by MM optimization on different parts of the amino acids,four types of RMSD are evaluated.The full,main chain,backbone,and side chain RMSDs refer to the RMSD on all heavy atoms,main chain heavy atoms,heavy atoms in the main chain except that in the termini,and the heavy atoms in the side chain of amino acids,respectively. The results are summarized in Table IV.
As shown in Table IV,the performances of ENCADS and GROMOS53A6 are clearly inferior to that of AMBER99SB,CHARMM27 and OPLSAA/L.It is also clear that the performance of AMBER99SB,CHARMM27 and OPLSAA/L for the capped amino acids is the poorest among the four terminus types of amino acids.This is unexpected as the capped species is used for deducing the force field parameters.A possible explanation may be that the force field parameters are determined by focusing on the dihedral angles and considering the solvent effect on the capped species. No matter what caused the results,however,it is fortunate to see that the three force fields may produce reasonable results for natural(neutral,protonated and deprotonated)amino acids in gas phase.
For natural amino acids,Table IV shows that the largest contribution to the RMSDs of the three force fields comes from the main chain.The RMSDs for the side chain and the backbone are fairly small.This means that the force fields are not very good at describing the termini of natural amino acids and the corresponding improvement is desirable.The three force fields perform similarly for the structures of neutral amino acids.CHARMM27 and OPLSAA/L also perform similarly for the structures of protonated and deprotonated amino acids.For capped amino acids,CHARMM27 is slightly better than OPLSAA/L and AMBER99SB.Overall,CHARMM27 performs the best,followed by OPLSAA/L.However,no significant differences are found among the three force fields of CHARMM27,OPLSAA/L,and AMBER99SB.
IV.CONCLUSION
The performances of five representative force fields on the structures and energies of neutral,protonated,deprotonated,and capped amino acids in gas phase have been analyzed systematically.AMBER99SB,CHARMM27 and OPLSAA/L are shown to clearly outperform GROMOS53A6 and ENCADS and the latter two force fields should not be considered for the study of gaseous amino acids and peptides.
AMBER99SB,CHARMM27,and OPLSAA/L perform reasonably well in terms of relative conformational energies,with AAEs all smaller than 2 kcal/mol. However,very large MAEs are also encountered so that care should be taken and verification of results should be conducted when possible.In terms of the H-bond description,AMBER99SB is slightly better than CHARMM27 and OPLSAA/L for neutral amino acids,while CHARMM27 performs well for capped,de-protonated and protonated amino acids.OPLSAA/L tends to overestimate the tendency of H-bond formation,but is the best for estimating the H-bond strength. In terms of structure prediction,CHARMM27 and OPLSAA/L are slightly better than AMBER99SB for neutral and capped amino acids.Both CHARMM27 and OPLSAA/L also perform well for predicting the conformations of protonated and deprotonated amino acids.Overall,CHARMM27 is the most recommended,followed by OPLSAA/L,for the study of gaseous amino acids and peptides,though it is noted that the performances of the three force fields are comparable when applicable.
V.ACKNOWLEDGMENTS
This work is supported by the National Natural Science Foundation of China(No.11374272),the National Basic Research Program of China(No.2012CB215405),and the Collaborative Innovation Center of Suzhou Nano Science and Technology.
[1]W.D.Cornell,P.Cieplak,C.I.Bayly,I.R.Gould,K. M.Merz,D.M.Ferguson,D.C.Spellmeyer,T.Fox,J. W.Caldwell,and P.A.Kollman,J.Am.Chem.Soc. 117,5179(1995).
[2]M.Karplus and D.L.Weaver,Nature 260,404(1976).
[3]D.J.Price and C.L.Brooks,J.Comput.Chem.23,1045(2002).
[4]A.D.Mackerell,J.Comput.Chem.25,1584(2004).
[5]A.Liwo,C.Czaplewski,S.OO ldziej,and H.A.Scheraga,Curr.Opin.Biotechnol.18,134(2008).
[6]K.A.Beauchamp,Y.S.Lin,R.Das,and V.S.Pande,J.Chem.Theory Comput.8,1409(2012).
[7]K.Lindorff-Larsen,P.Maragakis,S.Piana,M.P.Eastwood,R.O.Dror,and D.E.Shaw,Plos One 7,e32131(2012).
[8]D.A.Case,T.E.Cheatham,T.Darden,H.Gohlke,R.Luo,K.M.Merz,A.Onufriev,C.Simmerling,B. Wang,and R.J.Woods,J.Comput.Chem.26,1668(2005).
[9]B.R.Brooks,C.L.Brooks,A.D.MacKerell,L.Nilsson,R.J.Petrella,B.Roux,Y.Won,G.Archontis,C. Bartels,and S.Boresch,J.Comput.Chem.30,1545(2009).
[10]M.Levitt,M.Hirshberg,R.Sharon,and V.Daggett,Comput.Phys.Commun.91,215(1995).
[11]W.R.Scott,P.H.Hnenberger,I.G.Tironi,A.E.Mark,S.R.Billeter,J.Fennen,A.E.Torda,T.Huber,P. Krger,and W.F.van Gunsteren,J.Phys.Chem.A 103,3596(1999).
[12]W.L.Jorgensen,D.S.Maxwell,and J.Tirado-Rives,J.Am.Chem.Soc.118,11225(1996).
[13]J.W.Ponder and D.A.Case,Adv.Protein Chem.66,27(2003).
[14]K.Yang,B.Yang,and Z.Lin,Chin.J.Chem.Phys. 28,161(2015).
[15]H.Li,Z.Lin,and Y.Luo,Chem.Phys.Lett.610,303(2014).
[16]H.Li,Z.Lin,and Y.Luo,Chem.Phys.Lett.608,398(2014).
[17]R.Pang and Z.Lin,Chin.J.Chem.Phys.27,189(2014).
[18]H.Li,Z.Lin,and Y.Luo,Chem.Phys.Lett.598,86(2014).
[19]H.Li,Z.Lin,and Y.Luo,Chin.J.Chem.Phys.25,681(2012).
[20]H.Chen,Y.Wang,X.Chen,and Z.Lin,Chin.J.Chem. Phys.25,77(2012).
[21]A.D.Mackerell and N.K.Banavali,J.Comput.Chem. 21,(2000).
[22]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G. E.Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani,V.Barone,B.Mennucci,G.A.Petersson,H. Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F. Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa,M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai,T.Vreven,J.A.Montgomery,Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand,K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo,J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev,A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski,R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09:User’s Reference,Wallingford CT,USA:Gaussian Inc.105(2009).
[23]C.I.Bayly,P.Cieplak,W.Cornell,and P.A.Kollman,J.Phys.Chem.97,10269(1993).
[24]A.D.MacKerell,M.Feig,and C.L.Brooks,J.Comput. Chem.25,1400(2004).
[25]R.V.Devi,S.S.Sathya,and M.S.Coumar,Appl.Soft Comput.27,543(2015).
[26]W.Yu,L.Liang,Z.Lin,S.Ling,M.Haranczyk,and M.Gutowski,J.Comput.Chem.30,589(2009).
[27]V.Hornak,R.Abel,A.Okur,B.Strockbine,A.Roitberg,and C.Simmerling,Proteins:Struct.Funct. Bioinformatics 65,712(2006).
[28]C.Oostenbrink,A.Villa,A.E.Mark,and W.F.Van Gunsteren,J.Comput.Chem.25,1656(2004).
[29]M.Levitt,J.Mol.Biol.168,595(1983).
[30]G.A.Kaminski,R.A.Friesner,J.Tirado-Rives,and W.L.Jorgensen,J.Phys.Chem.B 105,6474(2001).
[31]W.Yu,Z.Wu,H.Chen,X.Liu,A.D.MacKerell Jr.,and Z.Lin,J.Phys.Chem.B 116,2269(2012).
[32]J.Benesty,J.Chen,Y.Huang,and I.Cohen,Pearson Correlation Coefficient,in:Noise Reduction in Speech Processing,Berlin:Springer,1-4(2009).
[33]C.L.Lawson and R.J.Hanson,Solving Least Squares Problems,SIAM,(1974).
[34]N.J.Nagelkerke,Biometrika 78,691(1991).
[35]J.E.Kerrigan,Biochemistry 32,13123(1993).
[36]B.Hess,D.van Der Spoel,and E.Lindahl,Netherland: University of Groningen,(2010).
∗Author to whom correspondence should be addressed.E-mail:zjlin@ustc.edu.cn,Tel:+86-551-63600345
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Incorporation of Reactive Corrosion Inhibitor in Waterborne Acrylic Polyurethane Coatings and Evaluation of its Corrosion Performance
- Response Ability to External Signal Enhanced by Biological Spatial Configuration in Coupled Hindmarsh-Rose Neural System
- Synthesis and Surface Activity of Heterogemini Imidazolium Surfactants
- Substituent Effects on Reduction Potentials of Meta-substituted and Para-substituted Benzylideneanilines
- Preparation of Nitrogen-Doped Carbon Catalyst to Oxygen Reduction Reaction and Influence of Protective Gas Flowing on Its Activity
- Structural and Magnetic Properties of Chemically Synthesized Pd-Modified NiFe2O4Nanoparticles