理论研究水和甲酸催化甲胺与羟基自由基的反应
2016-09-03陈广慧翟艳玲汕头大学理学院广东汕头55063山西师范大学化材学院山西临汾0400
王 爽,陈广慧*,张 祥,翟艳玲,陈 伟(.汕头大学理学院,广东 汕头 55063;2.山西师范大学化材学院,山西 临汾 0400)
理论研究水和甲酸催化甲胺与羟基自由基的反应
王爽1,陈广慧1*,张祥2*,翟艳玲1,陈伟1
(1.汕头大学理学院,广东汕头515063;2.山西师范大学化材学院,山西临汾041001)
本文采用量子化学计算方法和变分过渡态理论研究了水或甲酸的催化作用下羟基自由基提取甲胺中甲基氢和氨基氢的反应机理和动力学.在MC-QCISD//MP2/6-311++G(d,p)理论水平下计算的势能面表明,水或甲酸催化作用下的过渡态形成了很强的氢键,提取甲基氢反应过渡态的相对能量从裸反应的0.72 kcal·mol-1分别降低到-4.59和-9.78 kcal·mol-1;提取氨基氢的反应过渡态相对能量则从裸反应的-0.40 kcal·mol-1分别降低到-2.25和-9.12 kcal· mol-1.然而,提取甲基氢的速控步的能垒却从5.93 kcal·mol-1变为5.68和7.30 kcal·mol-1;提取氨基氢的能垒则从4.81 kcal·mol-1增加到8.04 kcal·mol-1和7.96 kcal·mol-1.但是,动力学计算表明298 K下水或甲酸催化下的反应速率常数分别比裸反应小3或2个数量级,而且考虑水或甲酸催化该反应前驱络合物的浓度后,计算得到的有效速率常数则分别降低6或8个数量级,因此水或甲酸均不能加速大气中甲胺与羟基的提氢反应.
CH3NH2+OH;水或甲酸催化;反应机理;速率常数
0 引言
脂肪胺可以反应生成氰化氢(HCN)和笑气(N2O),而这两种气体又是重要的温室气体[1]和平流层NOx[2-3]的重要原料,这使得脂肪胺在大气和环境科学中起着重要作用.氨基是生物燃料中的常见基团[4-5],在受到污染的对流层中羟基自由基是有机物降解产生的最常见活性物种之一.因此探究羟基与这些含氨基化合物的反应机理及速率,对于大气化学和光化学污染有重要意义.正因为此,脂肪胺与羟基自由基的反应被理论计算和实验广泛研究.例如,实验上:Atkinson等[6]通过瞬时光解共振荧光技术(flash photolysis-resonance fluorescence technique)研究了299~426 K下甲胺与羟基自由基的反应;Carl等[7]在富氢条件下用连续双光子解离NO2作为羟基自由基的来源,研究了295 K下甲胺、二甲胺、三甲胺以及乙胺与羟基自由基的反应.理论上:Galano和Alvarez-Idaboy[8]通过变分过渡态理论分析了甲胺、二甲胺以及乙胺与羟基自由基的不同反应路径.由于甲胺既可以从甲基中脱出氢,也可以从氨基中脱出氢生成两种自由基同分异构体CH2NH2和CH3NH.所以甲胺与羟基自由基可能的提氢反应有两种:
由于单个水分子可与羟基自由基或其他分子基团形成氢键,最近很多工作涉及到以水作为催化剂来降低有羟基自由基参与的反应的反应能垒[9-14].这些理论研究均表明单个水分子能够降低反应能垒,但是反应前期所形成的含水的前驱络合物的浓度对于总反应的速率常数至关重要.除水以外,甲酸分子也能降低异构化和水解反应的过渡态能量,Montu[15]发现甲酸能够将SO3的水解反应转变为无能垒过程.
本次工作将通过理论计算研究水或甲酸催化下CH3NH2+OH的提氢反应,通过势能面信息阐明催化反应的反应机理,并通过直接动力学计算判断水或甲酸是否能够加速大气中甲胺与羟基自由基的反应.
1 计算方法
所有的电子结构计算均在高斯09程序包[16]中进行,所有驻点(反应物、过渡态、复合物和产物)的结构优化和频率计算均采用MP2/6-311++G(d,p)方法.通过采用该方法和基组利用内禀反应坐标理论验证了各个过渡态所连接的反应物和产物,从而获得最低能量路径,同时计算了该路径下各结构的能量梯度和海森矩阵.各驻点的能量采用了被认为处理开壳层最有效的MC-QCISD方法进行了单点能校正[17].为了确保该水平的计算结果真实可信,MPW1K/6-311++G(d,p)和BB1K/6-311++G(d,p)方法也用于计算各驻点结构和频率,并用MC-QCISD方法进行单点能校正.通过对比可知,3种方法计算出的各驻点结构和采用MC-QCISD方法进行单点能校正后的结果十分相近,所以文中如无特别标注,采用的计算方法均为MP2/6-311++G(d,p)方法.
速率常数的计算采用Truhlar等人开发的Polyrate 2010程序[18].本文中变分过渡态理论采用带有小曲率隧道方法(small curvature tunneling,SCT)的正则变分过渡态理论(canonical variational transition-state theory,CVT).
2 结果与讨论
2.1无催化剂条件下CH3NH2与OH自由基的反应
为了研究H2O或FA对CH3NH2与OH自由基反应的影响,首先研究裸反应.前驱络合物CH3NH2…OH(R1)中,OH自由基的氢原子与CH3NH2的氮原子形成氢键(N…H—O),氢键大小为5.21 kcal·mol-1(图1,表1).从R1开始反应,有两个反应路径Ia和Ib(图2).路径Ia:经过TS1,OH自由基提取CH3NH2中NH2的一个氢原子形成后驱络合物P1,其中水分子中的氢原子和CH3NH自由基的氮原子形成氢键(N…H—O),P1很快分解为CH3NH和H2O;反应路径Ib:经过TS2,OH自由基提取CH3NH中CH3的一个氢原子,随后产生了后驱络合物P2.其中水分子的氢原子与CH2NH2的氮原子形成氢键(N…H—O),随后P2很快分解为CH2NH2和H2O.在MC-QCISD//MP2/6-311++G(d,p)理论水平下计算上述结构能量,TS1和TS2相对反应物的能量分别为0.72和-0.40 kcal·mol-1(图2),与Galano等[11]报道的0.97(TS1)和0.36 kcal·mol-1(TS2)很接近.后驱络合物P1和P2键能大小分别为4.25和3.05 kcal·mol-1.在298 K,反应路径Ia和Ib的反应焓变(ΔH)分别为-18.44和-26.75 kcal·mol-1,吉布斯自由能变(ΔG)分别为-16.61和-20.39 kcal·mol-1(表1),与Galano等[8]报道的-19.08和-24.48 kcal·mol-1很接近.我们的计算表明在裸反应中反应路径Ib具有较低的能垒,同时放出较多的热量,预示是该路径为主反应通道,这也可以在后续的速率常数计算中得到证实.从表1的数据可以看出,MC-QCISD//MPW1K 和MC-QCISD//BB1K水平计算的各类能量和参数都十分接近,与MC-QCISD//MP2的差值均小于1.00 kcal·mol-1.
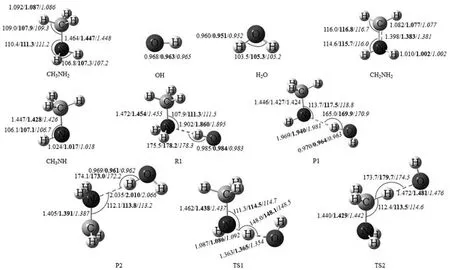
图1 在MP2/6-311++G(d,p)水平下优化CH3NH2+OH的反应物、产物、复合物和过渡态的几何构型.(键长:埃,键角:度)
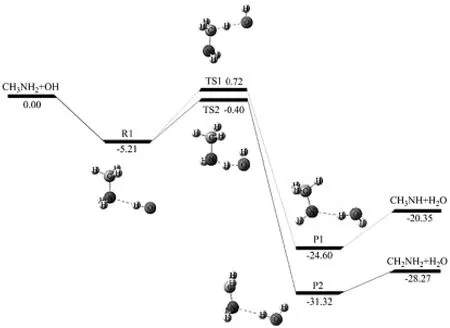
图2 在MC-QCISD//MP2/6-311++G(d,p)水平计算的CH3NH2+OH反应的势能面.
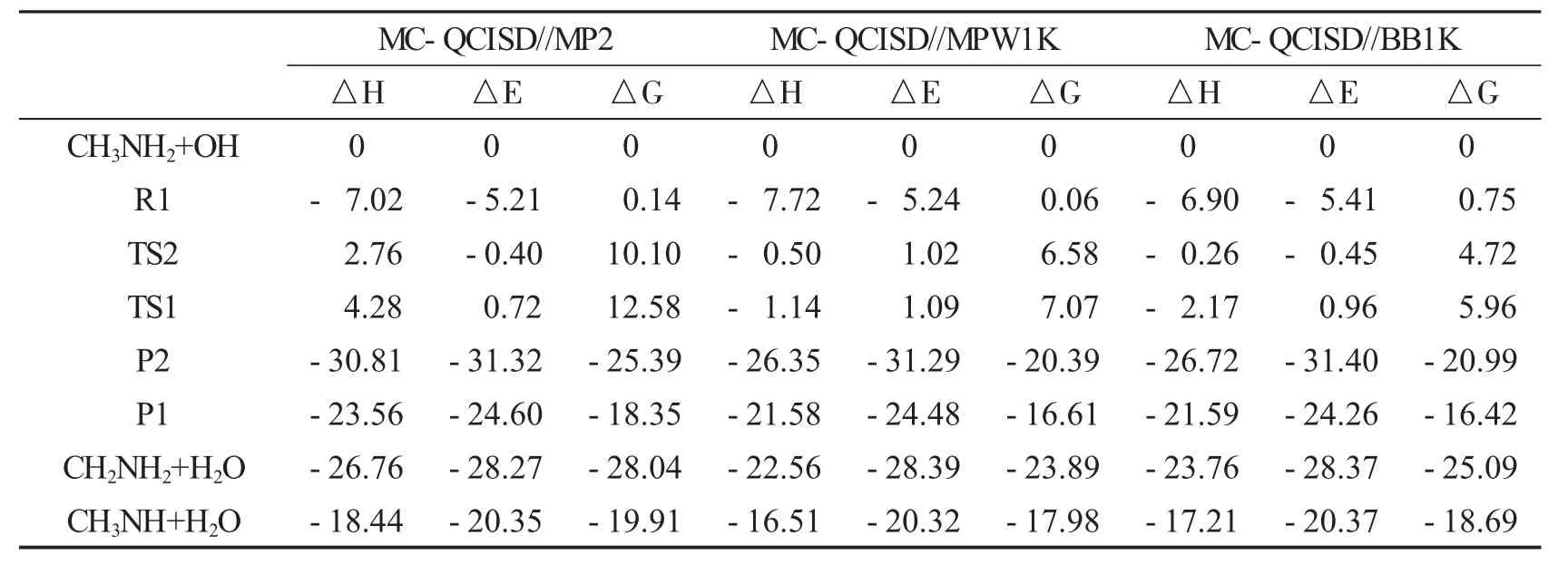
表1 在MC-QCISD//MP2、MC-QCISD//MPW1K和MC-QCISD//BB1K水平下计算298 K下反应CH3NH2+OH的焓变、零点能校正后的势垒和相对自由能. kcal·mol-1
2.2水催化CH3NH2与OH自由基的反应
在单个水分子存在时,三个分子直接反应的可能性比两分子相继反应的可能性小,所以该反应先形成一个两体络合物,然后与第三个物种发生碰撞形成三体络合物.三种两体络合物分别为CH3NH2…H2O、H2O…HO、CH3NH2…OH.为了确定这些二体络合物的稳定性,必须计算CH3NH2…OH(R1)、CH3NH2…H2O(R2)和H2O…HO(R3)的键能和平衡常数.裸反应中,CH3NH2…OH(R1)的键能是5.21 kcal·mol-1.CH3NH2…H2O(R2)中,CH3NH2的氮原子和H2O的氢原子形成氢键(N…H—O),键能大小为4.78 kcal·mol-1(图3,表2).H2O…HO(R3)中OH自由基的氢原子和水的氧原子形成氢键(O…H—O),键能大小为3.09 kcal·mol-1,在三个络合物中R3的键能最小(图3,表2).计算结果与Pesia Soloveichik[18]在CCSD(T)/CBS//B3LYP/aug-cc-pVTZ水平下计算得到的O…H—O氢键键能3.6 kcal·mol-1很相近.298 K下这些络合物的平衡常数分别为4.16×10-21,3.15× 10-21,5.55×10-22cm3·molecule-1(表3).在对流层,H2O、OH和CH3NH2的浓度分别为7.64×1017、1.00×106和1.00×1013molecules·cm-3[19],大气层中CH3NH2…H2O(R2)的浓度为2.41×1010molecules·cm-3,然而H2O…HO(R3),CH3NH2…OH(R1)浓度分别为4.24×102和4.16×10-2molecules·cm-3,络合物R1的浓度太低,所以不考虑包括R1的反应.以CH3NH2…H2O+OH和H2O…HO+CH3NH2为反应物,会形成两个相似的三体络合物R4和R5.IIa和IIb两个反应路径的方程式如下:
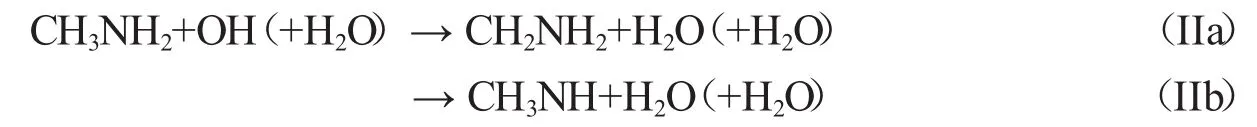
路径IIa:CH3NH2…H2O(R2)与OH反应或H2O···HO(R3)与CH3NH2反应,生成络合物R4(图4).R4是七元环结构,在R4中,H2O中氢原子与CH3NH2中的氮原子形成氢键(N…H—O),OH自由基中氢原子与H2O中的氧原子形成氢键(O…H—O),OH自由基的氧原子与CH3NH2中甲基氢原子形成一个弱氢键(C—H…O).R4相对于R2+OH的键能大小为5.49 kcal·mol-1,相对R3+CH3NH2键能大小为7.18 kcal·mol-1.R4经过TS3 (-4.00 kcal·mol-1),生成了六元环结构的后驱络合物P3,其中存在O…H—O、O…H—N、N…H—O三个氢键.作为催化剂水分子的氧原子与新形成水分子的氢原子形成氢键(O…H—O),新形成水分子的氧原子与CH2NH2中NH2的氢原子形成氢键(O…H—N),CH2NH2的氮原子与催化剂水分子的氢原子形成氢键(N…H—O).随后,P3分解产生CH2NH2和两个H2O.(R2 or R3→R4→TS3→P3→CH2NH2+2H2O)
路径IIb:CH3NH2…H2O(R2)与OH反应或H2O…HO(R3)与CH3NH2反应,生成络合物R5(图4).它是六元环结构,相对R2键能大小为5.51 kcal·mol-1,相对R3键能大小为7.20 kcal·mol-1(图4).R5经过TS4(-2.25 kcal·mol-1),生成产物P4,P4很快分解为CH3NH和两个H2O.(R2 or R3→R5→TS4→P4→CH3NH+2H2O)
在 298 K,H2O…HO(R3,4.24×102molecules·cm-3)的浓度比 CH3NH2…H2O (R2,2.41×1010molecules·cm-3)低(表3),而且键能最小(图4),因此以CH3NH2…H2O (R2)为二体络合物的反应路径是主要反应路径.
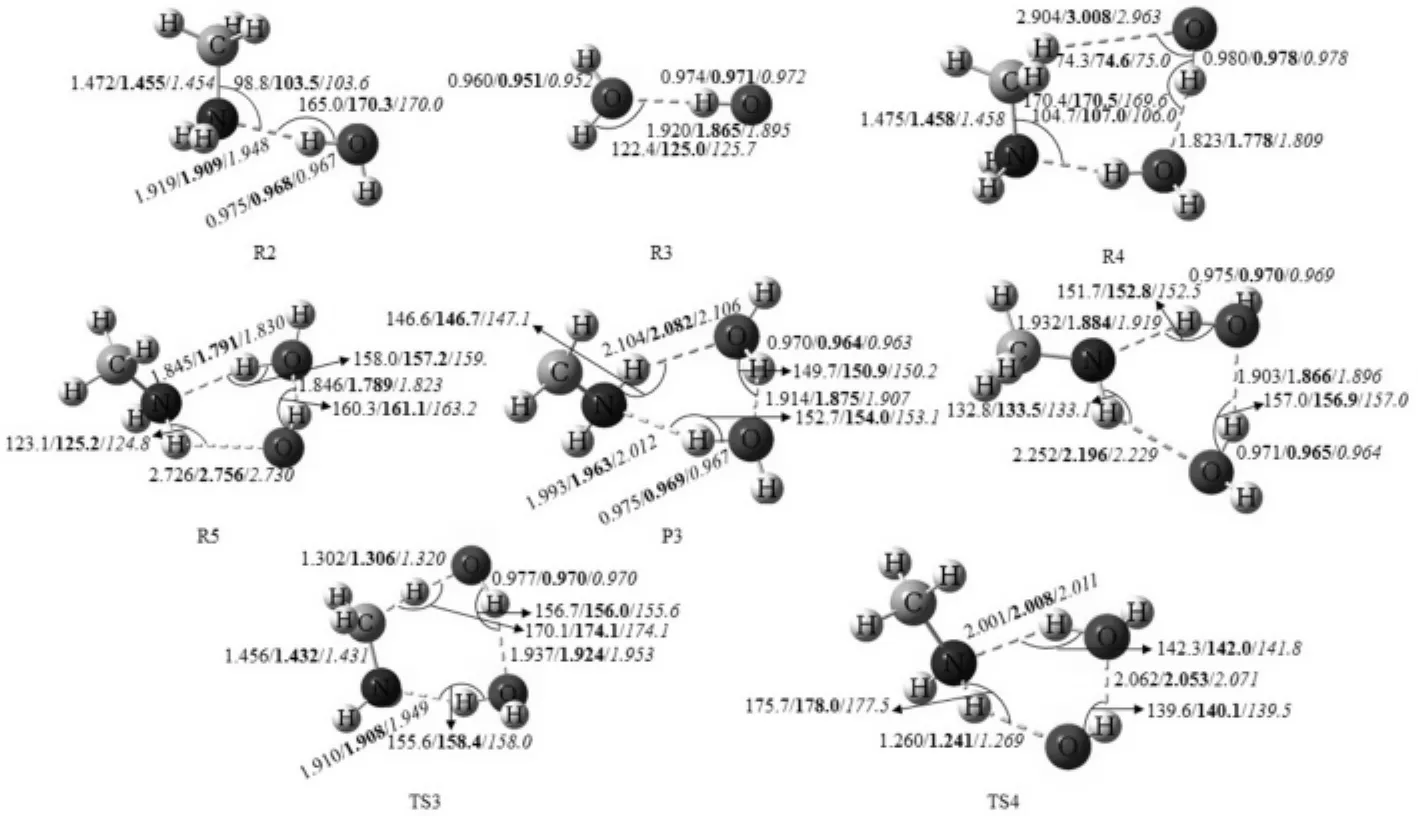
图3 在MP2/6-311++G(d,p)水平下优化CH3NH2+OH+H2O的反应物、产物、复合物和过渡态的几何构型.(键长:埃,键角:度)
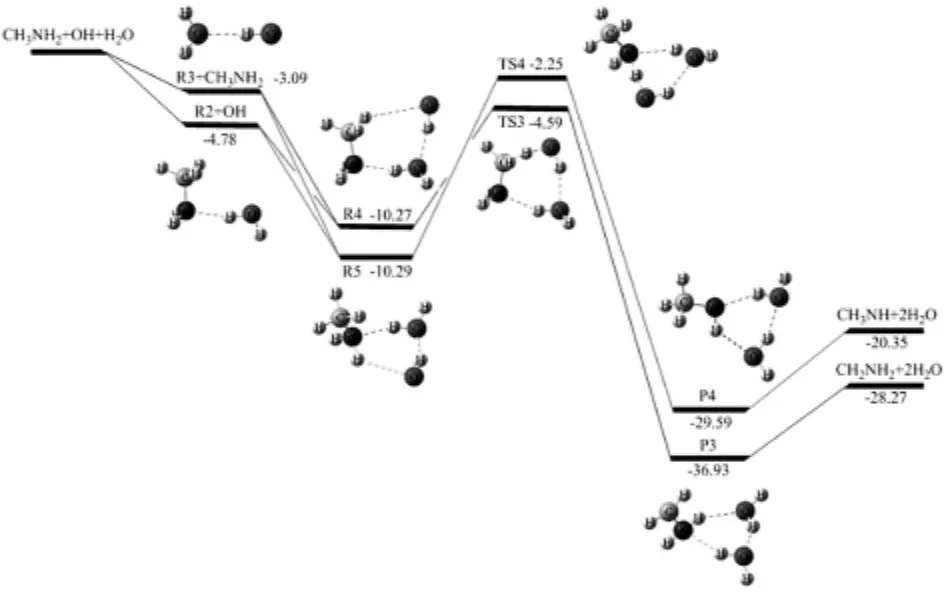
图4 在MC-QCISD//MP2/6-311++G(d,p)水平计算的CH3NH2+OH+H2O反应的势能面.
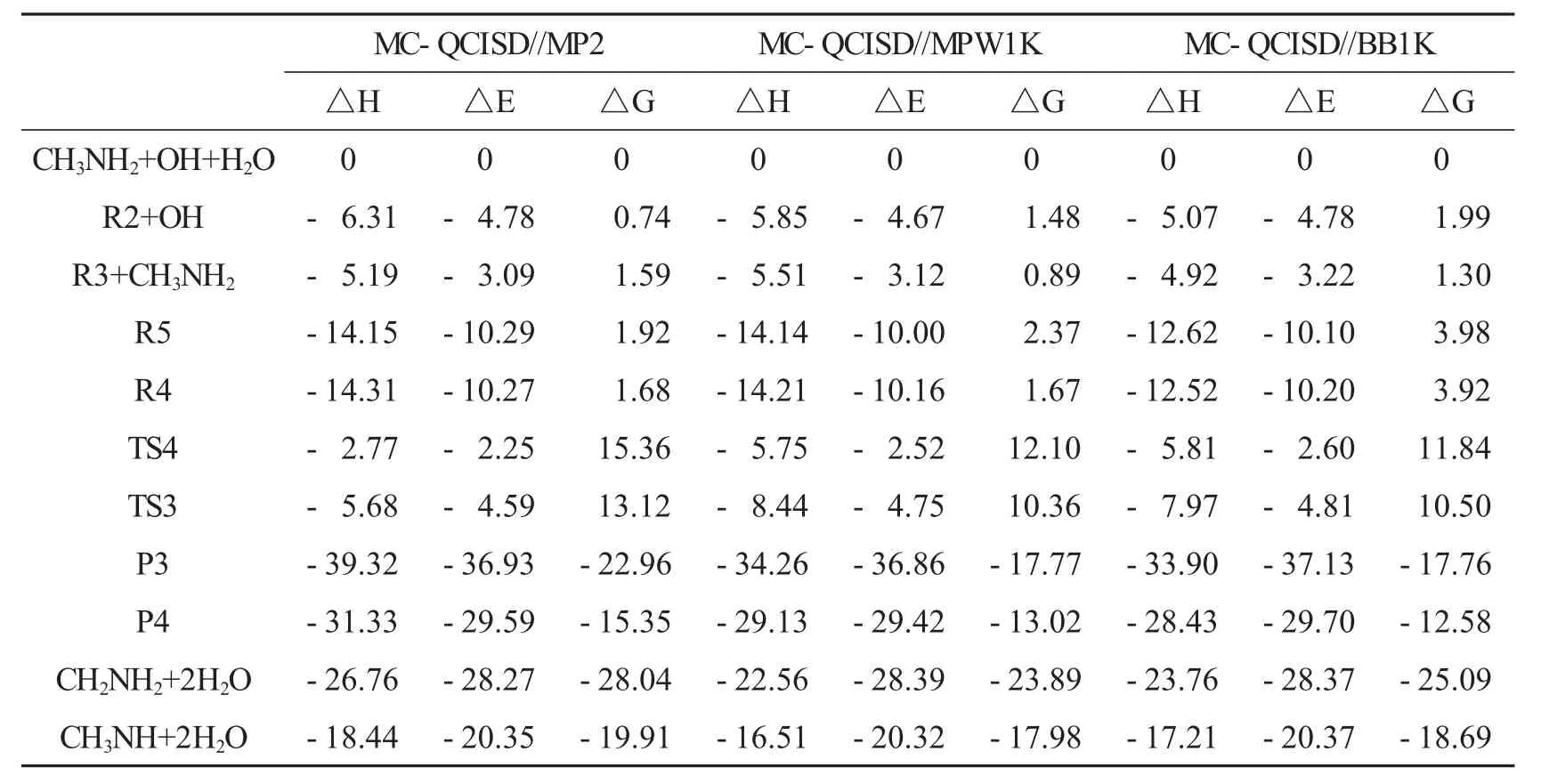
表2 在MC-QCISD//MP2、MC-QCISD//MPW1K和MC-QCISD//BB1K水平下计算298 K下反应CH3NH2+OH+H2O的焓变、零点能、校正后的势垒和相对自由能. kcal·mol-1
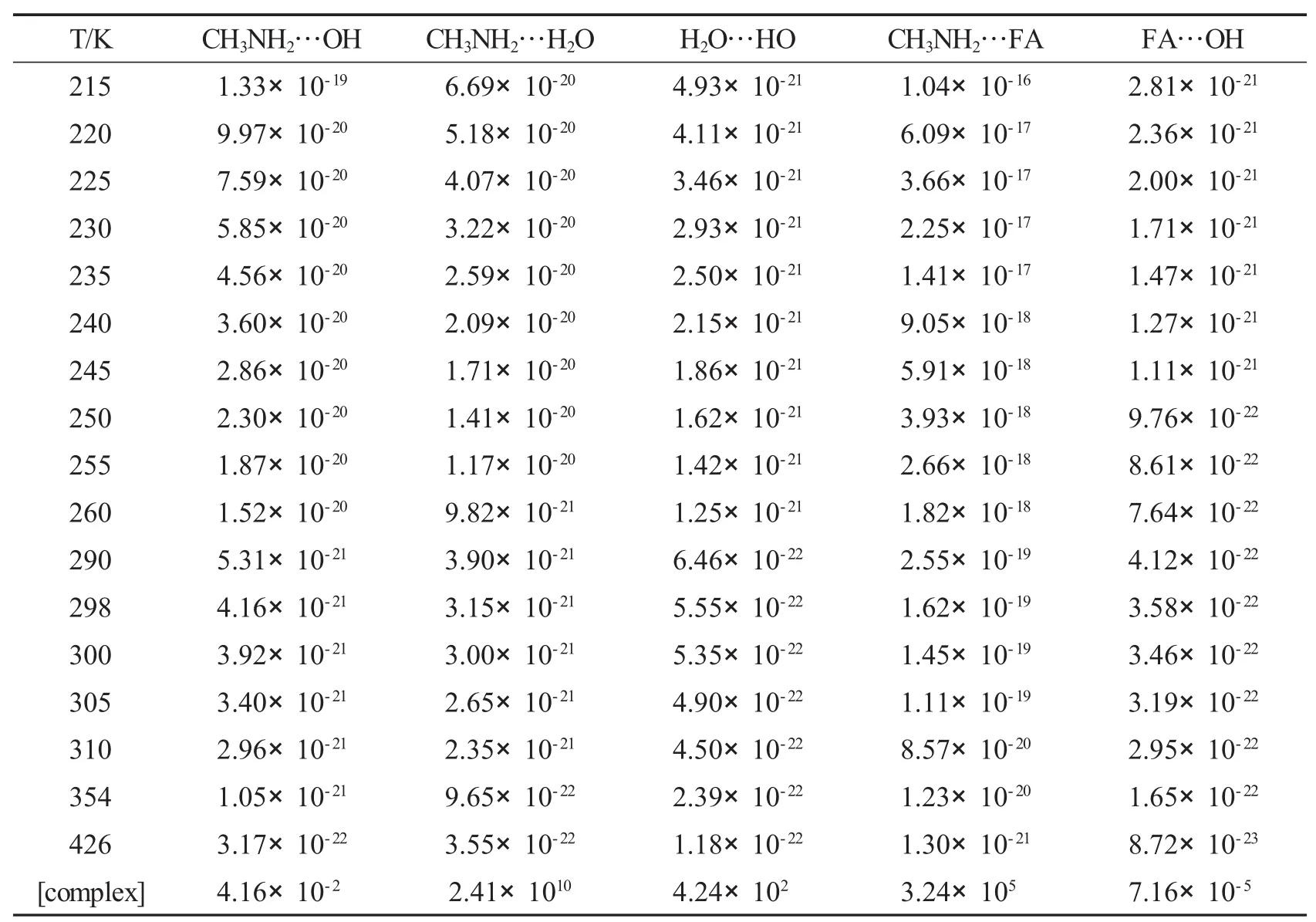
表3 在215-426 K下CH3NH2+OH反应相应二体络合物的平衡常数(cm3·molecule-1)及浓度(molecules·cm-3,298 K)
2.3甲酸催化CH3NH2与OH自由基的反应
甲酸催化CH3NH2与OH自由的反应和水催化该反应的方式相似.CH3NH2…FA (R6)OH或FA…OH(R7)+CH3NH2形成了络合物R8,R8是八元环结构(图5).R8中FA的氢原子与CH3NH2的氮原子形成氢键(N…H—O),OH自由基的氢原子与FA的氧原子形成氢键(O…H—O),OH自由基氧原子与CH3NH2中氨基氢原子形成氢键(N—H…O). R8相对R7+CH3NH2的键能为13.71 kcal·mol-1(图5,表4).
IIIa、IIIb反应路径的方程式如下:
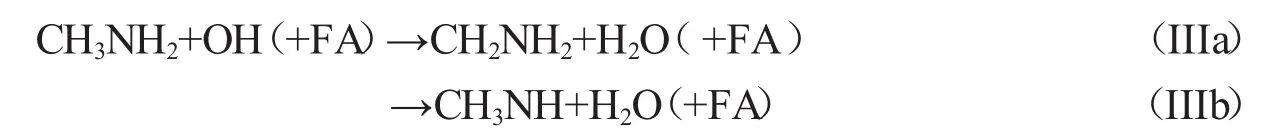
路径IIIa是CH3NH2…FA(R6)与OH自由基反应(图6).R8比R6+OH的能量高6.88 kcal·mol-1(表4),经过过渡态TS5(-9.0 kcal·mol-1),OH自由基提取CH3的H,生成后驱络合物P5.它是存在N—H…O、O—H…O、N…H—O三个氢键的八元环结构,NH2中的氢原子与新形成水分子中的氧原子形成氢键(N—H…O),新形成水分子的氢原子与FA的氧原子形成氢键(O—H…O),CH2NH2的氮原子与FA氢原子形成氢键(N…H—O). P5分解产生CH2NH2、H2O和FA.(R8→TS5→P5→CH2NH2+H2O+FA)
路径IIIb是OH自由基提取NH2中的氢原子(图6).R8经过TS6(-9.12 kcal·mol-1),生成P6,它是包括O—H…O、N—H…O、N…H—O三个氢键的八元环结构,NH2的氢原子和新形成水分子的氧原子形成氢键(N—H…O),新形成水分子的氢原子与FA的氧原子形成氢键,CH3NH的氮原子与FA的氢原子形成氢键(N…H—O).随后,P6很快分解产生CH3NH、H2O和FA.(R8→TS6→P6→CH3NH+H2O+FA)
298 K下FA…OH(R7,7.16×10-5molecules·cm-3)的浓度比CH3NH2…FA(R6,3.24× 105molecules·cm-3)低(表3),而且R7的键能最小(图6),因此以CH3NH2…FA(R6)为反应物的反应路径是主要反应路径.
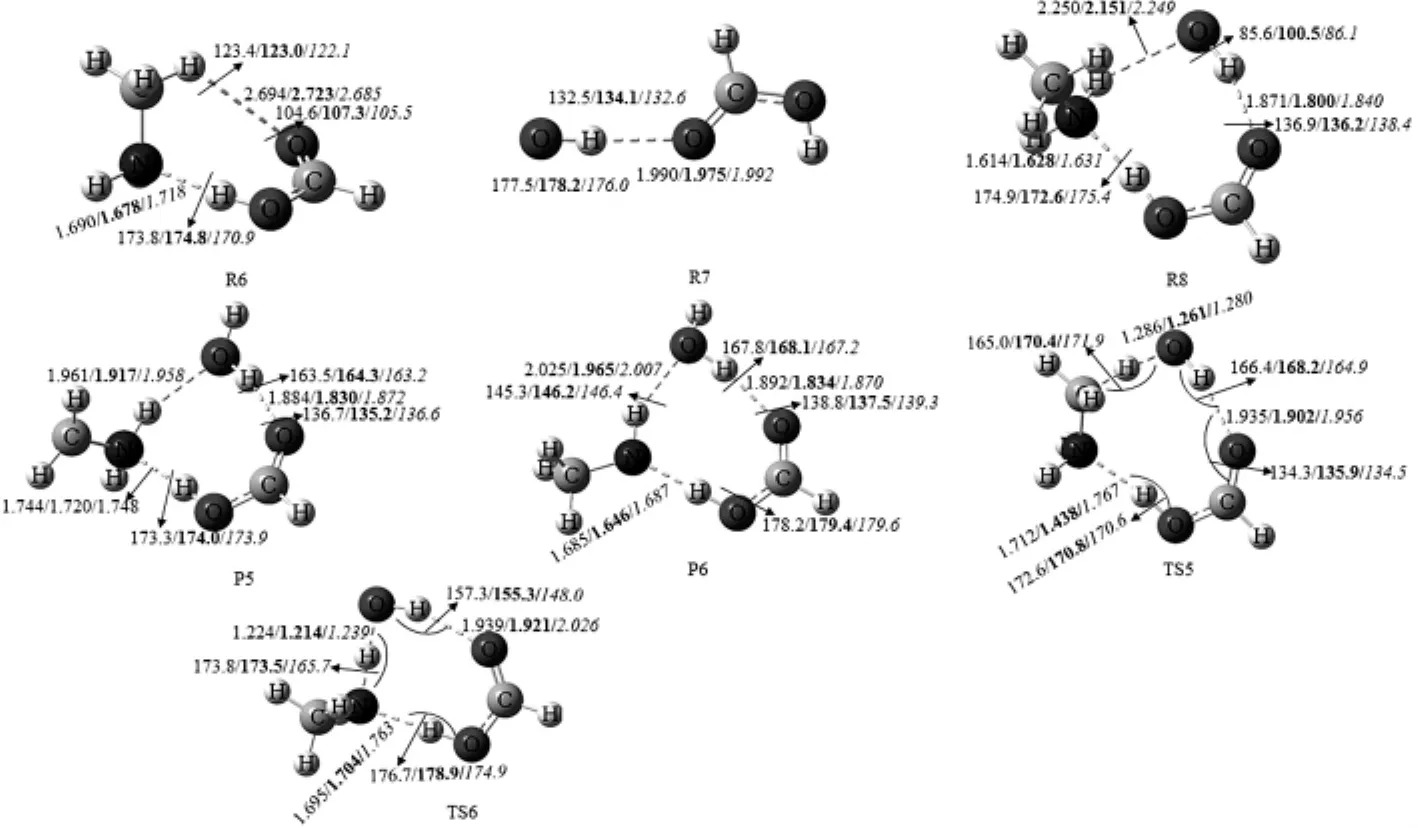
图5 在MP2/6-311++G(d,p)水平下优化CH3NH2+OH+FA的反应物、产物、复合物和过渡态的几何构型.(键长:埃,键角:度)
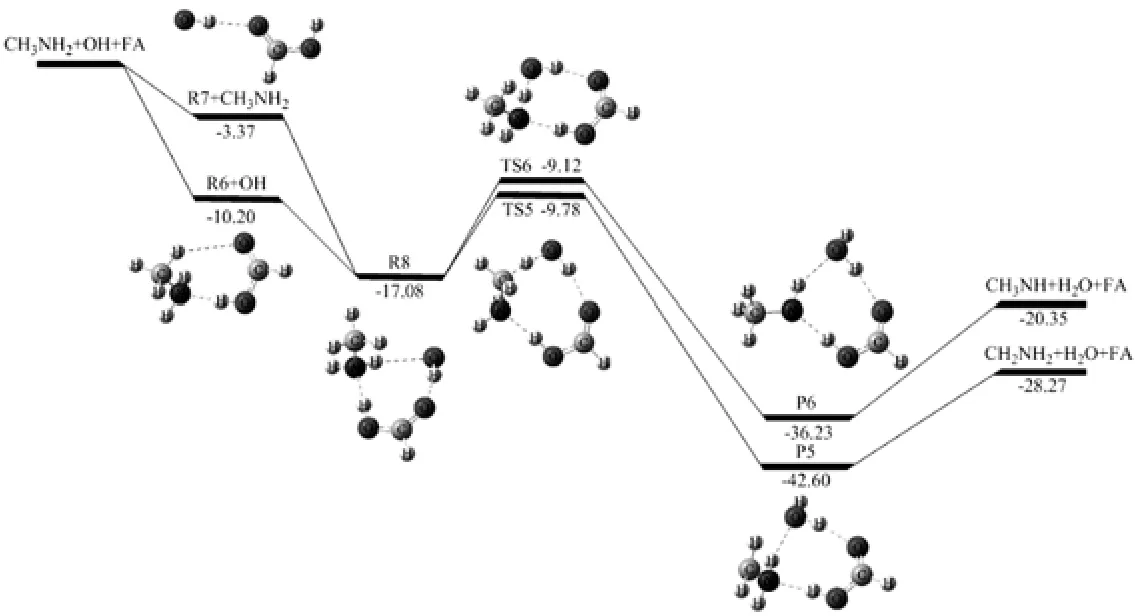
图6 在MC-QCISD//MP2/6-311++G(d,p)水平计算的CH3NH2+OH+FA反应的势能面.
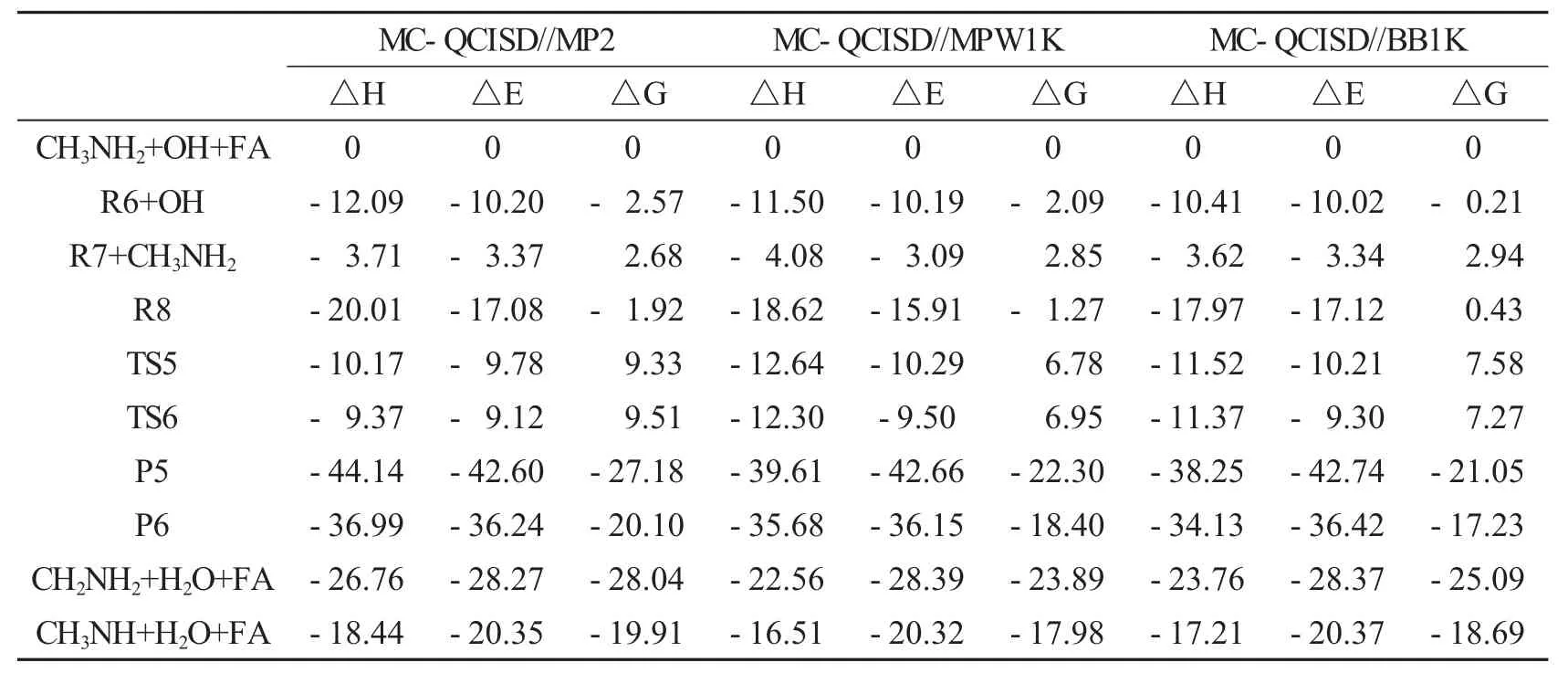
表4 在MC-QCISD//MP2、MC-QCISD//MPW1K和MC-QCISD//BB1K水平下计算298 K下反应CH3NH2+OH+FA的焓变、零点能、校正后的势垒和相对自由能. kcal·mol-1
2.4动力学和大气层的影响
在290-426 K,采用变分过渡态理论计算水或甲酸催化CH3NH2和OH自由基最可行反应路径的速率常数.
络合物反应的方程式如下:

速率常数的计算公式如下:

在高压限制条件下进行动力学模型修正,在此条件下前驱络合物通过和其他大气物种碰撞达到稳定.平衡常数Keq计算公式如公式(3);采用传统过渡态理论,速率常数k2的计算公式如(4)
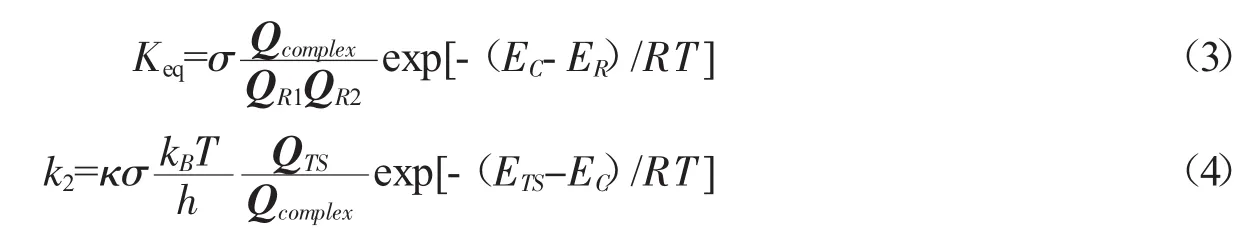
QTS、QR、Qc分别表示过渡态、反应物、前驱络合物的配分函数,kB为玻尔兹曼常数,h为普朗克常数,κ为穿透系数,σ为对称因子,ETS、ER、EC分别表示过渡态、反应物、络合物经过零点能校正的总能量.
采用变分过渡态理论(variational transition state theory,VTST)的计算公式如下:
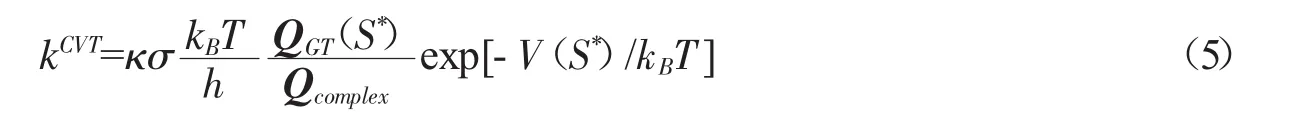
其中s*代表在温度T时沿反应路径自由能最大值,Qcomplex是前驱络合物的配分函数,QGT(s*)是广义过渡态的配分函数,V(s*)是势能,κ是采用小曲率计算的隧道效应参数.在MP2/6-311++G(d,p)水平计算配分函数,在MC-QCISD//MP2/6-311++G(d,p)水平计算单点能量,并进行零点能校正.
总速率常数的计算公式如下.

CH3NH2和OH自由基经过Ia和Ib两个反应路径的速率常数,如表5所示.在298 K,总速率常数为6.81×10-12cm3·molecule-1·s-1,与Annia Galano[8]提出的5.20×10-12cm3· molecule-1·s-1很相近.络合物CH3NH2…X(X=H2O,HCOOH)与OH自由基反应,速率常数分别比裸反应小0.001和0.1倍(表6,表7).

表5 在215-426 K下CH3NH2+OH反应的速率常数(cm3·molecule-1·s-1)

表6 在215-426 K下CH3NH2+OH+H2O反应的速率常数(cm3·molecule-1·s-1)

表7 在215-426 K下CH3NH2+OH+FA反应的速率常数(cm3·molecule-1·s-1)
为了探究变分效应和隧道效应对于速率常数计算的影响,以R1-Ib反应为例在图7中描述了反应通道R1-Ib的TST,CVT以及CVT/TST速率常数计算结果.从图中可以看出在整个温度区间CVT和CVT/TST,速率常数很接近,意味着隧道效应对R1-Ib反应通道的影响几乎可以忽略.在低温区TST和CVT速率常数极为接近,在高温区TST比CVT速率常数大.对于R1-Ib来说,500 K、1 500 K、2 000 K及2 400 K时kCVT/kTST的比值分别为0.90、0.36、0.29和0.25.因此,变分效应对R1-Ib反应通道的影响很小.
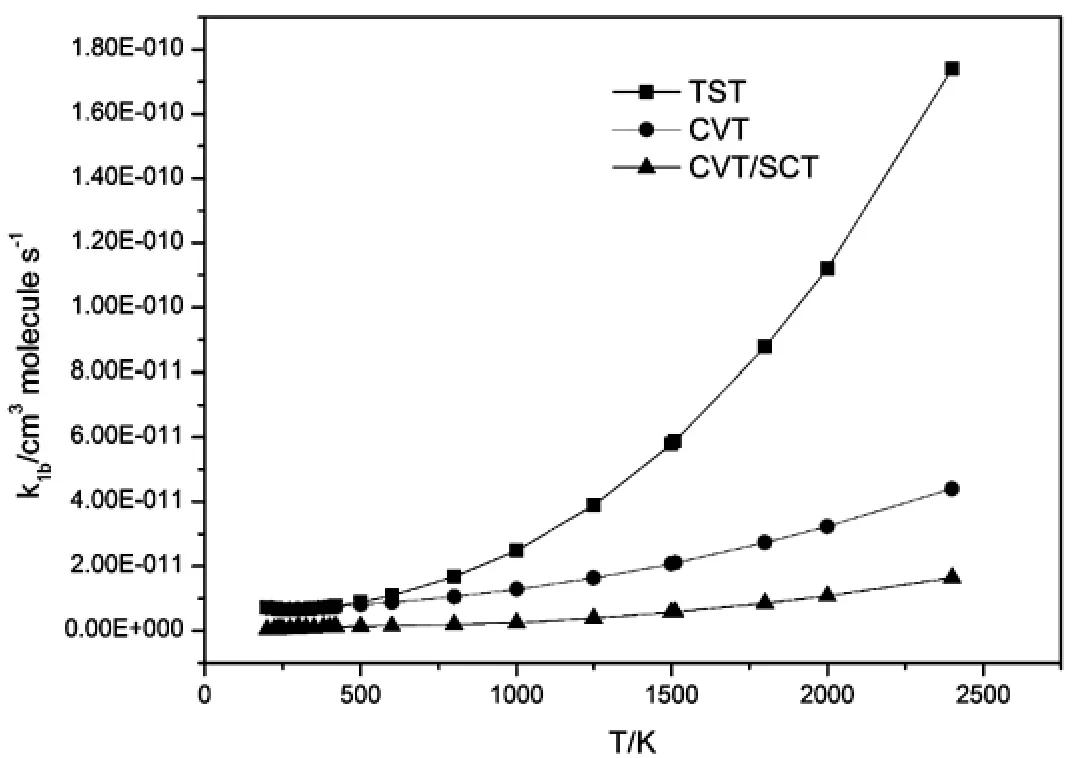
图7 反应通道R1-Ib在200~2 400 K温度区间内计算得到的TST,CVT以及CVT/TST的反应速率常数随温度变化的曲线.
为了获得水或FA催化CH3NH2和OH自由基反应更详细信息,有必要比较裸反应和有催化剂存在时各个反应的速率.
裸反应的速率计算公式如下:

催化剂存在时,速率计算公式如下:
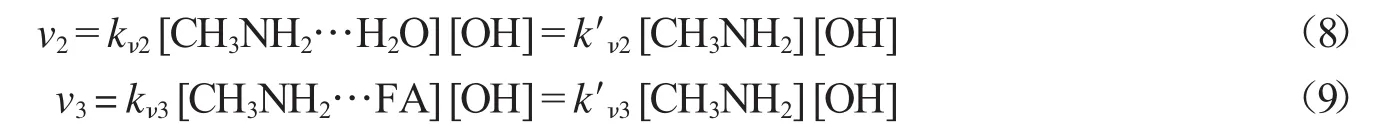
k′ν2=kν2Keq2[H2O]和k′ν3=kν3Keq3[FA],其中的Keq2与Keq3(表4)分别是形成络合物CH3NH2…H2O(R2)和CH3NH2…FA(R6)的平衡常数.k′ν2和k′ν3(表8)分别是由水和FA浓度决定的有效速率常数,它们可以直接与裸反应的速率常数kv1比较.298 K下CH3NH2…H2O与OH自由基反应的速率常数比裸反应小6个数量级,CH3NH2…FA与OH自由基反应则比裸反应小8个数量级.因为前驱双分子络合物的浓度比较低、催化剂存在时反应的能垒仍然比较高,所以水分子和FA减慢了反应速率.与此类似,单个水分子催化H2O2与OH自由基的反应,由于络合物H2O2…H2O的浓度比较低,反应能垒仍然比较大,导致水分子减慢了H2O2与OH自由基的反应速率[19].然而,水分子催化HCl与OH自由基反应,反应能垒低于裸反应的能垒,所以反应速率常数增大[20].同样,在气相,甲酸催化SO3的水解,由于甲酸的存在形成了一个无能垒的过程,所以甲酸加快了SO3的水解[15].通过比较水或FA催化CH3NH2与OH自由基反应的速率常数可知,水或FA均不能加快该反应的速率,因此推测,在大气中水和FA对CH3NH2的分解不重要.

表8 有、无催化剂时CH3NH2+OH反应的速率常数(k)和有效速率常数(k').cm3·molecule-1·s-1
3 结论
水和甲酸作为催化剂能明显降低甲胺与羟基自由基反应中对应过渡态的相对能量,但是其吉布斯自由能变却始终高于裸反应,而且动力学计算表明,298 K下催化反应路径的速率常数(水催化:5.31×10-15cm3·molecule-1·s-1;甲酸催化:3.52×10-14cm3·molecule-1·s-1)比裸反应路径(6.81×10-12cm3·molecule-1·s-1)的速率常数小.在考虑前驱络合物浓度时则该反应的有效速率常数比裸反应低6或8个数量级,这说明水和甲酸均不能加速大气中羟基自由基与甲胺的提氢反应.
[1]HOUGHTON J T,RASMUSSEN CALLANDER B A,VARNAY S K,et al.Intergovernmental Panel on Climatic Change(IPCC).Climatechange;The supplementary report to the IPCC scientific assessment[M]. NewYork:Cambridge University Press,1992.
[2]SCHADE G W,CRUTZEN P J.Emission of aliphatic amines from animal husbandry and their reactions:Potential source ofN2O and HCN[J].J Atmos Chem,1995,22(3):319-346.
[3]MCELROY MB,MCCONNELL J C.Nitrous oxide:A natural source of stratospheric NO[J].J Atmos Sci,1971,28(6):1095-1098.
[4]KOHSE-HÖINGHAUS K,OBWALD P,COOL T A,et al.Biofuelcombustion chemistry:from ethanol to biodiesel[J].AngewChem Int Ed,2010,49:3572-3597.
[5]LUCASSEN A,OBWALD P,STRUCKMEIER U,et al.Speciesidentification in a laminar premixed low-pressure flame of morpholine as a model substance for oxygenated nitrogen-containing fuels[J].Proc Combust Inst,2009,32(1):1269-1276.
[6]ATKINSON R,PERRY R A,PITTS J N.Rate constants for the reaction of the OH radical with CH3SH and CH3NH2over the temperature range 299-426 K[J].J Chem Phys,1977,66(4):1578-1581.
[7]CARL S,CROWLEY J.In the presence of H2as a source of OH inpulsed photolysis kinetic studies:rate constants for reaction ofOH with CH3NH2,(CH3)2NH,(CH3)3NH,and C2H5NH2[J].J Phys ChemA,1998,102(42):8131-8141.
[8]GALANO A,ALVAREZ-IDABOY J.Branching ratios of aliphatic amines+OH gas-phase reactions:a variational transition-state theory study[J].J Chem Theory Comput,2008,4(2):322-327.
[9]GONZALEZ J,ANGLADA J M.Gas phase reaction of nitric acid with hydroxyl radical without and with water.a theoretical investigation[J].J Phys Chem A,2010,114(34):9151-9162.
[10]BUSZEK R J,TORRENT-SUCARRAT M,ANGLADA J M,et al.Effects of a single water molecule on the OH+H2O2reaction[J].J Phys Chem A,2012,116(24):5821-5829.
[11]IUGA C,ALVAREZ-IDABOY J R,REYES L,et al.Can asingle water molecule really catalyze the acetaldehyde+OH reaction in tropospheric conditions?[J].J Phys ChemLett,2010,1(20):3112-3115.
[12]IUGA C,ALVZREZ-IDABOY J R,VIVIER-BUNGE A.Single water-molecule catalysis in the glyoxal+ OH reaction under tropospheric conditions:fact or fiction?a quantum chemistry and pseudo-second order computational kinetic study[J].Chem Phys Lett,2010,501(1):11-15.
[13]IUGA C,ALVZREZ-IDABOY J R,VIVIER-BUNGE A.On the possible catalytic role of a single water molecule in the acetone+OH gas phase reaction:a theoretical pseudo-second-order kinetics study[J]. Theor Chem Acc,2011,129(2):209-217.
[14]IUGA C,ALVZREZ-IDABOY J R,VIVIER-BUNGE A.Mechanism andkinetics of the water-assisted formicacid+OH reaction undertroposphericconditions[J].JPhysChemA,2011,115(20):5138-5146.
[15]HAZRA M K,SINHA A.Formic acid catalyzed hydrolysis of SO3in the gas phase:a barrierless mechanismfor sulfuric acid production ofpotential atmospheric importance[J].J AmChemSoc,2011,133 (43):17444-17453.
[16]FRISCH M J,TRUCKS G W,SCHEGEL H B,et al.Gaussian 09,revision A.02;Gaussian,Inc.:Wallingford,CT,2009.
[17]FAST P L,TRUHLAR D G.MC-QCISD:multi-coefficient correlation method based on quadratic configuration interaction with single and double excitations[J].J Phys Chem A,2000,104(26):6111-6116.
[18]CORCHADO J C,CHUANG YY,FASTP L,et al.POLYRATE-version 9.3 ed.UniversityofMinnesota,Minneapolis,2002.
[19]BUSZEK R J,TORRENT-SUCARRAT M,ANGLADA J M,et al.Effects of a single water molecule on the OH+H2O2reaction[J].J Phys Chem A,2012,116(24):5821-5829.
[20]BUSZED R J,BARKER J R,Francisco J S.Water effect on the OH+HC lreaction[J].J Phys Chem A,2012,116(19):4712-4719.
Catalytic Effect of Water and Formic Acid on the Reaction of CH3NH2with OH Radicals
WANG Shuang1,CHEN Guanghui1,ZHANG Xiang2,ZHAI Yanling1,CHEN Wei1
(1.Department of Chemistry,Shantou University,Shantou 515063,Guangdong,China 2.School of Chemistryand Material Science,Shanxi Normal University,Linfen 04100,Shanxi,China)
The reaction mechanisms and kinetics for the hydrogen abstraction reaction ofCH3NH2by OH radicals assisted by water,or formic acid(FA)have been investigated theoretically using quantum chemistry calculations and variational transition state theory.The potential energy surfaces(PESs)calculated at the MC-QCISD//MP2/6-311++G(d,p)levels oftheoryreveal that the relative energies to reactants of the transition states involving catalysts are significantly reduced (for the H abstraction of CH3:-4.59 kcal·mol-1with water,-9.78 kcal·mol-1with FA;for the H abstraction of NH2:-2.25 kcal·mol-1with water,-9.12 kcal·mol-1with FA)compared with that ofreaction without catalyst(for the H abstraction of CH3:0.72 kcal·mol-1;for the H abstraction of NH2:-0.40 kcal·mol-1)due to the formation of strong hydrogen bond(s).However,the barrier heights of rate-controlled step are increased to 8.04,5.68 kcal·mol-1(with water,for the H abstraction of NH2,CH3,respectively)and 7.96,7.30 kcal·mol-1(with FA,for the H abstraction of NH2,CH3,respectively)from 4.81,5.93 kcal·mol-1(for the H abstraction of NH2,CH3,respectively).The kinetics calculations show that the rate constants for water,formic acid assisted reactions are smaller than that“bare”reaction by about 3 or 2 orders of magnitude at 298 K,respectively.Furthermore,after taking into account the concentration of the prereactive bimolecular complexes with water and FA,the effective rate constants of reaction actually slow down by 6 or 8 orders of magnitude,respectively.Consequently,neither water nor formic acid can accelerate the title reaction in atmosphere.
CH3NH2+OH;water or FA;reaction mechanism;rate constants
O643.12
A
1001-4217(2016)01-0025-13
2015-10-23
陈广慧(1972—),男(汉),博士,教授,硕士生导师,主要从事理论化学计算方面研究.
E-mail:ghchen@stu.edu.cn.
张祥(1970—),男(汉),博士,副教授,硕士生导师,主要从事理论化学计算方面研究.
E-mail:xiangzh2000@hotmail.com.
广东省自然科学基金资助项目(s2013010014476);汕头大学国家自然科学基金培育项目资助(NFC13001)
