利用密度泛函理论研究苄硫醇在石墨烯表面的吸附机理
2016-08-09周乃武赵永驰王位丽张利萍
王 群,周乃武,赵永驰,王位丽,张利萍
(绵阳师范学院 生命科学与技术学院,四川 绵阳 621006)
0 引言
苄硫醇是一种具有令人窒息气味的剧毒物质,可以应用在很多领域,如医药、农药、橡胶再生剂及合成有机添加剂等[1-3].当人体吸入或通过皮肤吸收巯基类物质时会使人中毒;因此,在工业和住宅环境中监测它们的存在是非常有必要的.但由于其毒性较大,通过实验手段获得足够的硫醇类物质的有用信息具有相当大的挑战,计算模拟的方法可以弥补这方面的缺陷,而且可以在原子、分子水平上提供一些有价值的信息.
目前,采用计算模拟的方法对硫醇类物质已经开展了一些研究.例如,QIN等[4]利用DFT方法探讨了乙硫醇(C2H5SH)在纯的和缺陷石墨烯表面的吸附机理.SOSCUN等[5]和HU等[6]也用DFT方法分别研究了C2H5SH与GaN和沸石的相互作用.JU等[7]采用蒙特卡罗模拟探讨了C2H5SH与MFI和MOR沸石的详细吸附机理.VOLMER等[8]研究了C10H21SH在Fe表面的电化学性质及电子光谱.GREATHOUSE等[9]研究发现CH3SH,C2H5SH,C3H7SH可以吸附在石墨表面.
石墨烯由于其独特的结构、巨大的比表面积等特性在气体传感器方面使用比较广泛[10-11],目前也吸引了越来越多科学家们的研究兴趣.然而,石墨烯吸收气体主要是通过物理吸附,相互作用很弱[12],因此,科学家们在石墨烯中引入各种缺陷和金属掺杂来改善石墨烯的特性.石墨烯中掺杂导致了电子态的局域化,减少了电荷迁移率从而使掺杂位点化学反应活性提高,同掺杂类似,石墨烯中引入晶格缺陷也会导致接近狄拉克点的电子态局域化,从而也提高了化学反应活性[13-14].最近,对于石墨烯掺杂和引入缺陷的研究也取得了一些有价值的成果.例如,WANNO等[15]发现CO对Fe、Ru、Rh、Ni、Pd、Ir和Pt掺杂的石墨烯比纯的石墨烯更敏感.ZHANG等[16]发现H2S与Fe掺杂石墨烯比纯的石墨烯有更强的相互作用,他们也表明缺陷石墨烯和Ag掺杂的石墨烯表面比纯石墨烯表面对2,3,7,8-tetrochlorodibenzo-p-dioxins具有更大的吸附能力[17].虽然纯的、缺陷的和金属掺杂的石墨烯可以作为气体传感器,有效检测有害气体,但C7H7SH的吸附特性目前尚未被详细研究.本文采用DFT方法探讨了纯的、缺陷的及Hg/Pd掺杂的石墨烯对C7H7SH的吸附能力.通过吸附能,态密度,差分电荷密度详细地分析了它们之间的吸附机制.
1 模拟过程
1.1 模型的建立
苄硫醇和石墨烯模型: C7H7SH包含了一个苯环,一个亚甲基(-CH2),一个巯基(-SH),它的三维结构如图1a所示,结构优化之后S-C键长为0.1852 nm,苯环中C-C键长为0.1399 nm.纯的石墨烯模型是通过石墨切面获得,构建了一个包含50个碳原子的石墨烯晶胞, 表面大小为1.2300 nm×2.1304 nm.赋予石墨烯周期性,并在其上方加上厚度为2.5 nm的真空层,优化之后的C-C键长大约为0.1420 nm[18],如图1b所示.

图1 (a)苄硫醇的三维结构;(b)石墨烯的三维结构d1,d2,d3是石墨烯优化之后的键长.Fig. 1 The three-dimension structures of (a) C7H7SH;(b)G
缺陷和Hg/Pd掺杂石墨烯模型:缺陷石墨烯模型是在纯石墨烯模型上去掉一个碳原子,Hg/Pd掺杂的石墨烯模型是用金属原子取代一个碳原子得到,优化后的几何模型展示在图2a-2c中.在缺陷石墨烯中由于相邻的一些碳原子的移除,导致了它们的键长最小的减少到0.1375 nm,如图2a.而在Hg掺杂的石墨烯里面,靠近Hg原子的C-C键长在0.1341 nm到0.1493 nm之间变化,如图2b.在Pd掺杂的石墨烯里面,靠近Pd原子的C-C键长在0.1365~0.1446 nm之间变化,如图2c.

图2 (a)缺陷石墨烯三维结构;(b)Hg掺杂石墨烯三维结构;(c)Pd掺杂石墨烯三维结构d1,d2,d3分别是Hg/Pd掺杂石墨烯优化之后metal-C之间的距离Fig. 2 The three-dimension structures of (a)defective G;(b)Hg doped G (b) Pd doped G
苄硫醇在纯的、缺陷的、Hg/Pd掺杂的石墨烯上的吸附模型: 情况1-苄硫醇平躺在各个石墨烯表面,优化之后模型如图3a-3d所示.情况2-苄硫醇垂直放在各个石墨烯表面,且巯基靠近石墨烯表面,优化之后如图4a-4d.
1.2 模型参数
本文使用Materials Studio(Accelrys, San Diego, CA)中基于第一性原理DFT的Dmol3软件进行计算研究.该模块中用数值基组表示物理波函数,可以带来比较精确的结果,而且计算成本相对较低,采用等同于Gaussian中6-31G**基组的DNP双重数值基组(DNP double numerical basis set)[19-21].用DFT半核赝势(DFT semicore pseudopotentials)处理内层电子.用Perdew-Burke-Ernzerhof (PBE) 广义梯度近似(GGA)计算交换关联能[22].根据Monkhorst-Pack方案, 布里渊区域(Brillouin zone)内的K点网格设置为2×2×1[23], 费米拖尾效应(Fermi smearing)值设置为0.005 Ha(1 Ha=27.2114 eV), 全局轨道截断(global orbital cutoff)值设置为0.50 nm.体系的几何构型优化和能量计算收敛条件为:(a)自洽循环数量级≤1.0×10-6Ha/atom;(b)能量值数量级≤1.0×10-5Ha/atom;(c)最大应力≤0.02 Ha/nm;(d)最大位移≤0.0005 nm.

图3 情况1-结构优化后: (a) C7H7SH平躺在纯的石墨烯表面; (b) C7H7SH平躺在缺陷石墨烯表面; (c) C7H7SH平躺在Hg掺杂的石墨烯表面; (d) C7H7SH平躺在Pd掺杂的石墨烯表面Fig. 3 Case 1: After structure optimization

图4 情况2-结构优化后: (a) C7H7SH垂直放在纯的石墨烯表面; (b) C7H7SH垂直放在缺陷石墨烯表面;(c) C7H7SH垂直放在Hg掺杂的石墨烯表面; (d) C7H7SH垂直放在Pd掺杂的石墨烯表面Fig. 4 Case 2: After structure optimization
1.3 吸附能
各种吸附体系都在寻找最稳定结构,即寻找最低的吸附能,吸附能Eads定义为:
Eads=EC7H7SH+surface-EC7H7SH-Esurface.
(1)
此时,EC7H7SH+surface、EC7H7SH和Esurface分别代表总体系、C7H7SH和各个石墨烯表面的吸附能,本文中吸附能的值如表1所示,吸附能越负,则表示吸附体系越稳定.
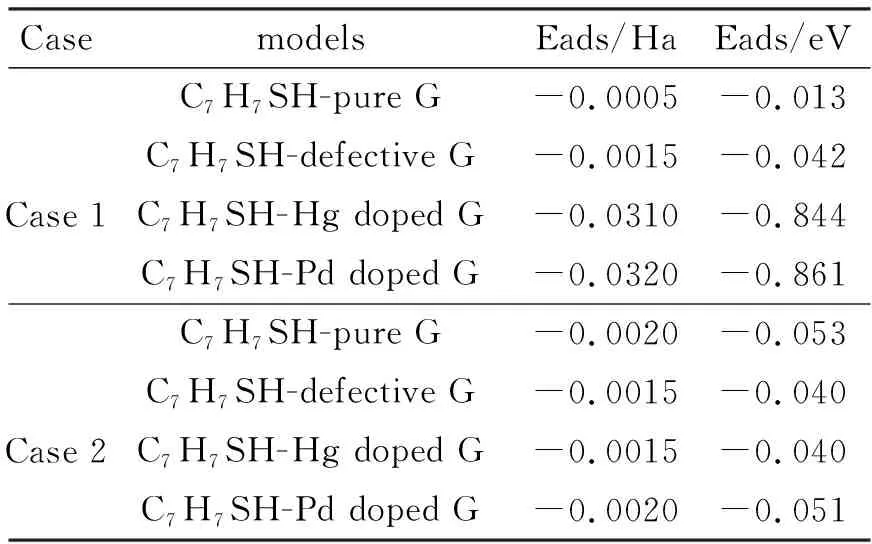
表1 C7H7SH吸附在纯的、缺陷的、Hg/Pd掺杂的石墨烯表面的吸附能Tab. 1 The adsorption energies of C7H7SH on the pure, defective, Hg/Pd-doped G surfaces
2 结果与讨论
2.1 Hg/Pd掺杂石墨烯几何结构优化后的稳定性分析

表2 几何结构优化之后C-C, Hg-C和Pd-C的距离Tab. 2 The distances of C-C, Pd-C and Hg-C after the structure optimization
2.2 吸附能的比较分析
各种体系的吸附能的值如表1所示,吸附能值越负表示这些吸附体系热力学上越稳定.在情况1中,C7H7SH吸附在纯的石墨烯表面,吸附能为-0.013 eV,表明它们之间有非常小的吸附作用(图3a),但是当C7H7SH吸附在缺陷的和Hg/Pd掺杂的石墨烯表面时,它们的吸附能分别是-0.042、-0.844和-0.861 eV,表明它们之间相比于C7H7SH与纯的石墨烯之间有更强的相互作用.缺陷石墨烯上悬挂的C键比较活跃,能够与吸附的分子发生相互作用,从而增强了缺陷石墨烯和分子的吸附,这一结论也是和GUO等[18]的结论一致,他们发现RGD能够更强的吸附在缺陷石墨烯上而不是纯的石墨烯上.Hg/Pd掺杂的石墨烯模型里面,结构优化之后Hg-S和Pd-S的距离分别是0.2555和0.2541 nm,表明掺杂金属和C7H7SH中S原子可以发生相互作用,此外,在C7H7SH中苯环的π电子也可以与石墨烯中π电子发生π…π相互作用.但是在C7H7SH中S原子的孤对电子和石墨烯的π电子之间还存在着强烈的排斥作用,最后由于这几个不同作用的共同影响的结果导致了C7H7SH和掺杂石墨烯之间的作用也不是太强.
为了与情况1比较,建立了情况2中的模型,在情况2中,C7H7SH的巯基靠近平面,而且整个C7H7SH与石墨烯表面垂直.研究发现所有模型的吸附能都相当低(-0.040~-0.053 eV),那是因为在情况2中,C7H7SH中-SH靠近石墨烯表面,S原子的孤对电子和石墨烯的π电子之间的排斥力占主导作用,如图4c-4d所示,Hg/Pd掺杂的石墨烯表面向C7H7SH相反的方向被排斥到了平面之外.但当排斥力最终被π…π相互作用弥补时,这些原子达到了最后的平衡态.这些结论与Qin等[4]的结论完全吻合,他们研究了C2H5SH在纯的、Stone-Wales 和vacancy缺陷的石墨烯表面的吸附,发现C2H5SH的巯基与石墨烯之间有大的排斥作用,导致了它们之间的相互作用都很弱.
2.3 态密度的分析
基于能带理论的态密度(density of states, DOS)表示某个能量状态下电子态的数目.如果相邻原子的局域态密度在同一个能量上同时出现了尖峰,则将其称之为杂化峰(hybridized peak),它可以直观地展示相邻原子之间的作用强弱.在接下来的分析中,主要用情况1中最稳定的两个掺杂模型进一步分析C7H7SH和掺杂石墨烯之间相互作用情况.从图5a和5b中可以看到,C7H7SH与Hg掺杂的石墨烯在-2.79、-3.19、-5.97、-6.15、-13.1 2 eV位置有峰重叠,C7H7SH与Pd掺杂的石墨烯在-2.82、-4.95、-6.19、-13.11、-14.29 eV之间有峰重叠,说明吸附质C7H7SH与石墨烯基底之间在这些能量所处的轨道上有两两作用的可能性.
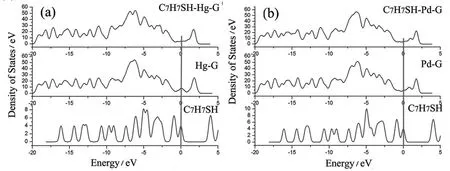
图5 情况1中C7H7SH-G体系的态密度图 (a) C7H7SH-Hg doped G; (b) C7H7SH-Pd doped GFig. 5 DOSs of the C7H7SH-G system(a) C7H7SH-Hg doped G; (b) C7H7SH-Pd doped G in the Case 1
2.4 差分电荷密度分析
电荷密度的结果进一步展示了C7H7SH和石墨烯之间的相互作用,图6 和 图7表明了情况1中C7H7SH平躺在石墨烯表面的总电荷密度和差分电荷密度的结果.差分电荷密度表明了电荷密度在吸附过程中的变化,是由整个吸附体系的总电荷密度(ρC7H7SH+surface)减掉C7H7SH (ρC7H7SH)和石墨烯表面(ρsurface)的电荷密度之和,公式如下:
△ρ=ρC7H7SH+surface-(ρC7H7SH+ρsurface).
(2)
浅色区域代表电子减少的区域,深色区域代表聚集的区域.观察总的电荷密度(图 6),发现在纯的、缺陷的石墨烯模型相比与掺杂的模型几乎没有电子重叠,表明C7H7SH与纯的,缺陷的石墨烯之间的相互作用很小.同时,通过差分电荷密度分析,在图7b、7c 和7d中发现电荷主要聚集在C7H7SH中的-SH周围,而在掺杂的体系中,-SH的S原子周围比缺陷体系中聚集更多电子,代表S原子得到了更多电子,金属原子周围是蓝色,代表金属原子失去了电子,通过差分电荷密度分析可以发现-SH中S原子与金属可以形成较强的相互作用.
同时对情况1中的C7H7SH和金属掺杂石墨烯的表面原子电荷密度进行二维切片,可以进一步看出在哪些原子轨道可以成键,如图8a-8b,从图8b可以很明显地看到-SH周围有电子聚集,Pd周围有电子减少,所以在Pd和S之间很明显地看到它们之间有相互作用,而图8a中,电子的聚集和减少相比于图8b不是太明显,这也可以很直观地说明为什么Pd掺杂石墨烯对C7H7SH的吸附比Hg掺杂石墨烯对C7H7SH吸附强.

图6 在情况1中不同的等值面图的总电荷密度
(a) C7H7SH-pure G; (b) C7H7SH-defective G;(c) C7H7SH-Hg doped G; (d) C7H7SH-Pd doped G
Fig. 6IsosurfaceoftotalelectrondensityfortheC7H7SH-GsystemwithdifferentisovaluesintheCase1:
(a)C7H7SH-pureG;(b)C7H7SH-defectiveG; (c)C7H7SH-HgdopedG;(d)C7H7SH-PddopedG
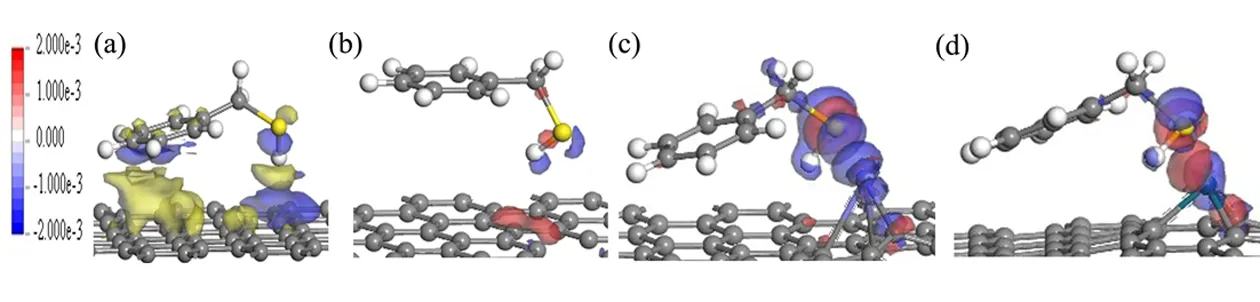
图7 在情况1中不同等值面的差分电荷密度 (a) C7H7SH-pure G; (b)C7H7SH-defective G; (c) C7H7SH-Hg doped G; (d) C7H7SH-Pd doped GFig. 7 Isosurface of difference electron density for C7H7SH-G systems with different isovalues in the Case 1 (a) C7H7SH-pure G; (b) C7H7SH-defective G; (c) C7H7SH-Hg doped G; (d) C7H7SH-Pd doped G

图8 在情况1中等值面为20 e/nm3的电子二维切片图 (a) C7H7SH-Hg doped G; (b) C7H7SH-Pd doped G
3 结论
研究有害的硫醇类物质在材料表面的吸附机制对于去除这些有毒的化学物质起着至关重要的作用.本研究调研了C7H7SH在纯的、缺陷的、Hg/Pd掺杂的石墨烯表面的吸附机理,主要的结论如下:(Ⅰ) 各种石墨烯表面的吸附能力一定程度上取决于C7H7SH的初始构型,C7H7SH平躺在石墨烯表面比垂直放在石墨烯表面有更大的吸附能力,同时在平躺的模型中,它们之间的相互作用是几种不同作用共同影响的结果.(Ⅱ) 对于平躺在石墨烯表面的模型,可以发现C7H7SH在掺杂石墨烯表面是最稳定的吸附构型,缺陷次之,纯的最不稳定.情况2中C7H7SH与石墨烯体系相互作用的强度是Pd掺杂石墨烯>Hg掺杂石墨烯>缺陷石墨烯>纯的石墨烯.希望这些结论能够对发展巯基类有害物质的石墨烯传感器提供有价值的理论参考.
