高通量测序分析20株禽流感病毒全基因
2016-08-09王楷宬庄青叶张笑春侯广宇王素春
王楷宬,庄青叶,张笑春,邱 源,王 通,侯广宇,刘 朔,王素春
(中国动物卫生与流行病学中心,青岛 266032)
高通量测序分析20株禽流感病毒全基因
王楷宬*,庄青叶,张笑春,邱源,王通,侯广宇,刘朔,王素春
(中国动物卫生与流行病学中心,青岛 266032)
摘要:为探索使用高通量测序测定和分析禽流感病毒全基因的方法,使用Ion Torrent PGM测序仪对20株自华东地区分离的禽流感病毒进行全基因测序,分析该方法的优缺点,以及此20株病毒的基因组特性和进化特征。结果显示,应用Ion Torrent PGM测序仪能够测出全部20株病毒的全基因,确定其均为H9亚型h9.4.2.5分支的低致病性毒株,NA基因均为N2亚型,并分析了其6个内部基因分子演化关系。说明高通量测序可用于禽流感病毒全基因分析。
关键词:高通量测序;禽流感;全基因;分子演化
禽流行性感冒病毒(avian influenza virus,AIV)简称禽流感病毒,属于正黏病毒科、甲型流感病毒属。甲型流感病毒粒子表面有血凝素(hemagglutinin,HA)和神经氨酸酶(neuraminidase,NA)两种表面结构蛋白,根据HA和NA的抗原性差异可分别将其分为18种H亚型(H1~H18)和11种N亚型(N1~N11)[1-3]。AIV在家禽和水禽中广泛存在,引起禽类的全身性或呼吸系统性疾病[4]。鸡、火鸡、鸭和鹌鹑等家禽及野鸟均可感染,且发病情况不一,有的急性死亡,有的感染后无明显症状。
AIV是单股负链RNA病毒,其基因组由8个单股负链RNA片段组成,每个基因在3′末端和5′末端都带有12~13个保守核苷酸序列。这8个片段可编码10种蛋白质,其中8种蛋白质(HA、NA、PB1、PB2、NP、M1、M2和PA)为结构蛋白,NS1和NS2为非结构蛋白,位于宿主细胞的细胞质中[5]。低保真RNA聚合酶可引起流感病毒的高突变率,以及基因重组,均可使流感病毒呈现分子多样性,使每个病毒亚型可进化为多个分支[6]。通常一个碱基对的突变,就可引起病毒对宿主感染能力的改变,造成对抗流感药物的抗性[7]。
由于AIV的突变率高,并易发生重组,监测其进化趋势和流行态势尤为重要。AIV所有节段的变化均可通过高通量测序方法进行检测。目前,研究人员已建立了在一个RT-PCR反应中使用一对通用引物扩增甲型流感病毒所有8个基因节段的方法,这使得AIV全基因组的测序更为简单[8]。传统的一代测序虽然经济高效,但仅能测定特异性扩增的某一片段,且不能测定准种[9]。随着高通量测序技术的发展,一些测序仪平台已能够进行甲型流感病毒全基因组的序列测定与分析,通过一次扩增AIV所有8个节段的RT-PCR反应,能够消除临床样品中来自宿主和环境中核酸的干扰,这使得利用高通量测序进行病毒核酸序列分析更为高效经济。另外,在文库构建过程中,可以给每份样品加入不同的索引标签,在同一个测序反应中就可以对数十甚至上千AIV的全基因进行测序,其经济性更为明显。
作者使用Ion Torrent PGM测序平台对未知亚型的AIV临床分离株进行全基因组测序,并分析其亚型和各节段基因序列的分子进化关系,为高通量测序技术用于AIV的大规模测序与分析奠定基础。
1材料与方法
1.1材料和仪器
PathAmp FluA Reagent Kit、Ion Xpress Plus Fragment Library Kit、Ion PGM Template OT2 200 Kit、Ion PGM Sequencing 200 Kit V2、E-Gel SizeSelect 2% Agarose、Ion 318 Chip Kit V2、 Dynabeads MyOne Streptavidin C1 Beads、Ion Xpress Barcode Aaptors、Qubit核酸浓度测定仪及配套试剂均购自Life technologies公司;QIAxtractor 高通量核酸提取仪及配套试剂盒(cador®Pathogen 96 QIAcube®HT kit);QIAgility 全自动体系构建系统及QIAxcel 全自动实时毛细管电泳仪及配套试剂盒(德国QIAGEN);超净工作台(美国Forma Scientific);移液器(Eppendorf);孵化器(德州诚信孵化设备有限公司);高速台式离心机(德国Heraeus Biofuge primoR);PCR扩增仪(Perkin Elmeter Gen Amp PCR System 9600)。
1.2毒株
20株禽流感病毒由中国动物卫生与流行病学中心禽病监测室保存,2014年7月分离自华东地区活禽交易市场的拭子样品和粪便样品,均按农业行业标准(NY/ T 772-2013)的特异性M基因引物检测为禽流感病毒,但未确定亚型。毒株名称见表1。
1.3RNA提取
按照说明书操作,使用QIAxtractor高通量核酸提取仪(德国QIAGEN)及其配套试剂盒(Cador®Pathogen 96 QIAcube®HT kit),提取病毒RNA。
1.4流感病毒全基因扩增
应用PathAmp FluA Reagent Kit对病毒 RNA进行反转录,并扩增生成全基因组cDNA。使用Qubit核酸浓度测定仪及配套试剂测定DNA浓度。
1.5文库构建
纯化后的AIV全基因cDNA均稀释成35 μL中含有200 ng DNA的溶液,使用Ion Xpress Plus Fragment Library Kit将病毒全基因组DNA打断为200 bp的片段,并分别加入含有Barcode的接头,构建为PGM测序仪可以识别的DNA文库。
1.6测序
将20个DNA文库分别稀释为26 pmol·L-1,并等量混合。应用Ion PGM Template OT2 200 Kit对混合的DNA文库进行测序前的样品处理。处理后的样品加样至Ion 318 芯片,置于PGM测序仪进行测序。测序序列经PGM自带的FluAnalysis(v4.0)软件进行序列拼接和亚型分析。
1.7分析与毒力或宿主嗜性相关的关键位点
使用Lasergene软件将20株病毒的HA基因翻译为蛋白质序列,分析HA蛋白的裂解位点(第333—341位氨基酸)序列[10],分析NS蛋白中结合CPSF的特异性残基是否为GLEWN[11]。
1.8分子进化分析
利用NCBI的Influenza Virus Resource数据库,搜索并下载1966—2014年其收录的来自中国的H9亚型AIV的全基因序列,HA基因与136株从NCBI 流感数据库下载的序列进行比对与进化分析,NA基因与86株从NCBI 流感数据库下载的序列进行比对与进化分析。在分析本论文所用20株分离株的6个内部基因时,除添加从NCBI 流感数据库下载的86株序列,另添加经Blast比对与该基因同源性较高的几个毒株(H7、H10、H9N9以及mixed亚型)的序列,分别与测序得到的序列一起进行在线Alignment初步对比,用MUSCLE方法进行对齐运算[12-13]。利用MEGA6.0生物信息学软件,应用Bayesian反馈信息准则(Bayesian Information Criterion,BIC)的替代模式和系统评分,构建进化树。通过Find Best DNA/Protein Model运算方法,计算得出构建进化树的最佳运算模型,利用Construct/Test Maximum Likelihood Tree运算模型,运算Bootstrap值为1 000,绘制系统进化树。谱系按照WHO/OIE/FAO联合发布的方法进行划分和命名[14]。
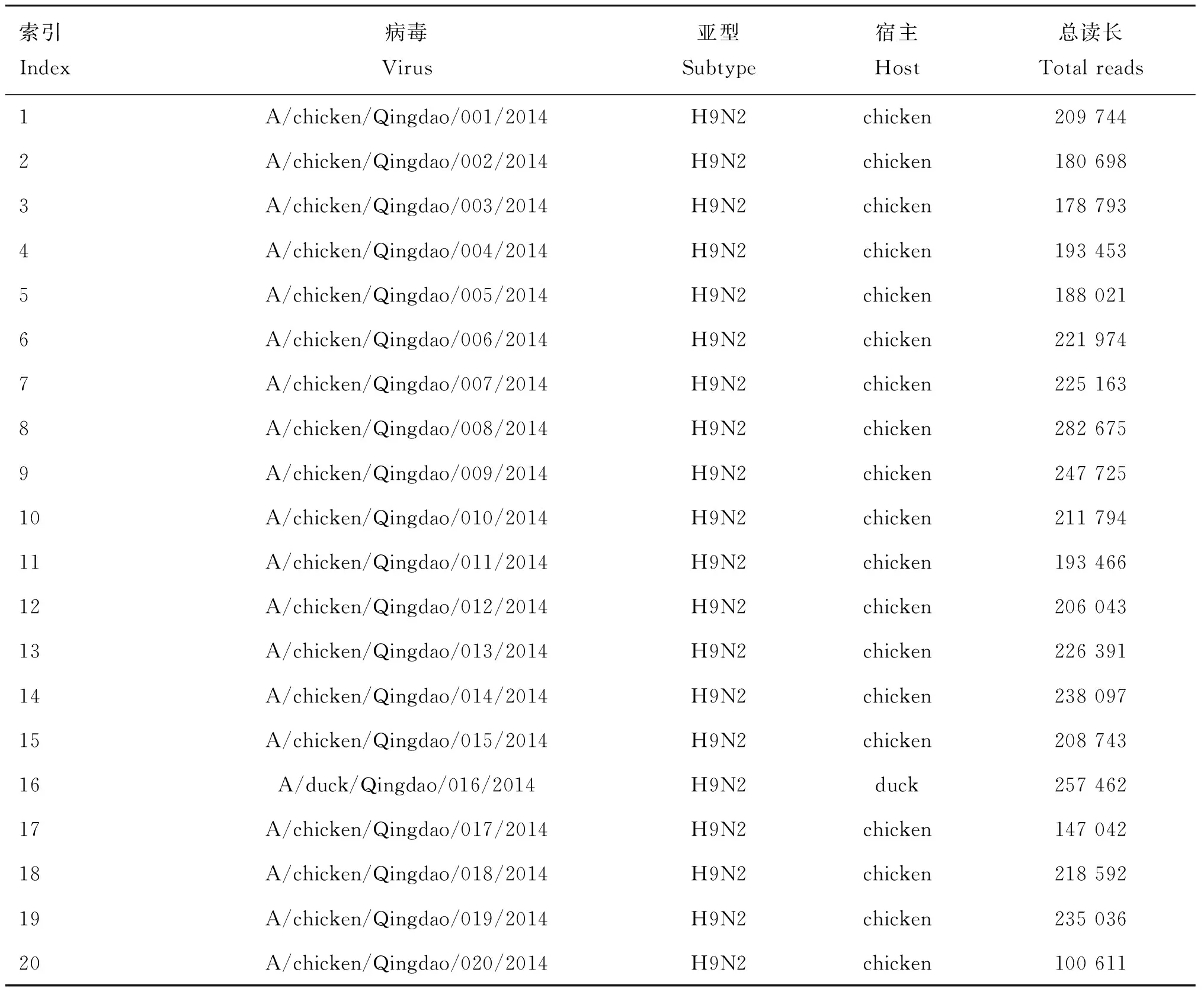
表1 毒株名称、亚型、宿主与序列数量
2结果
2.1测序质量
测序数据已上传至GenBank,登录号为SRR2083856。测序共产生7 950 401个reads,去除接头序列和低质量序列后,4 171 523个reads含有1~20 barcode中的一个,其中3 157 806个reads正确匹配到甲型流感病毒的序列上,涵盖了所有20个AIV毒株的所有节段。每个AIV毒株各节段的测序reads数量见表1。
2.2测序深度
AIV毒株每个节段的平均测序深度见表2。包括各节段的5′和3′端序列,所有的节段至少平均被测372次(PB1基因)。M基因的平均测序深度最大,为2 449.9。
2.3与毒力或宿主嗜性相关的关键位点
所有20株AIV的HA蛋白的裂解位点(第333—341位氨基酸)序列均为PSRSSR↓GLF,为低致病性毒株[10]。NS1 蛋白能结合CPSF,其结合CPSF的特异性残基为GLEWN。分析发现,所测20个分离株特异性残基中的L均突变为F,即GLEWN 突变为GFEWN,或可导致这些毒株致病力增强[11]。
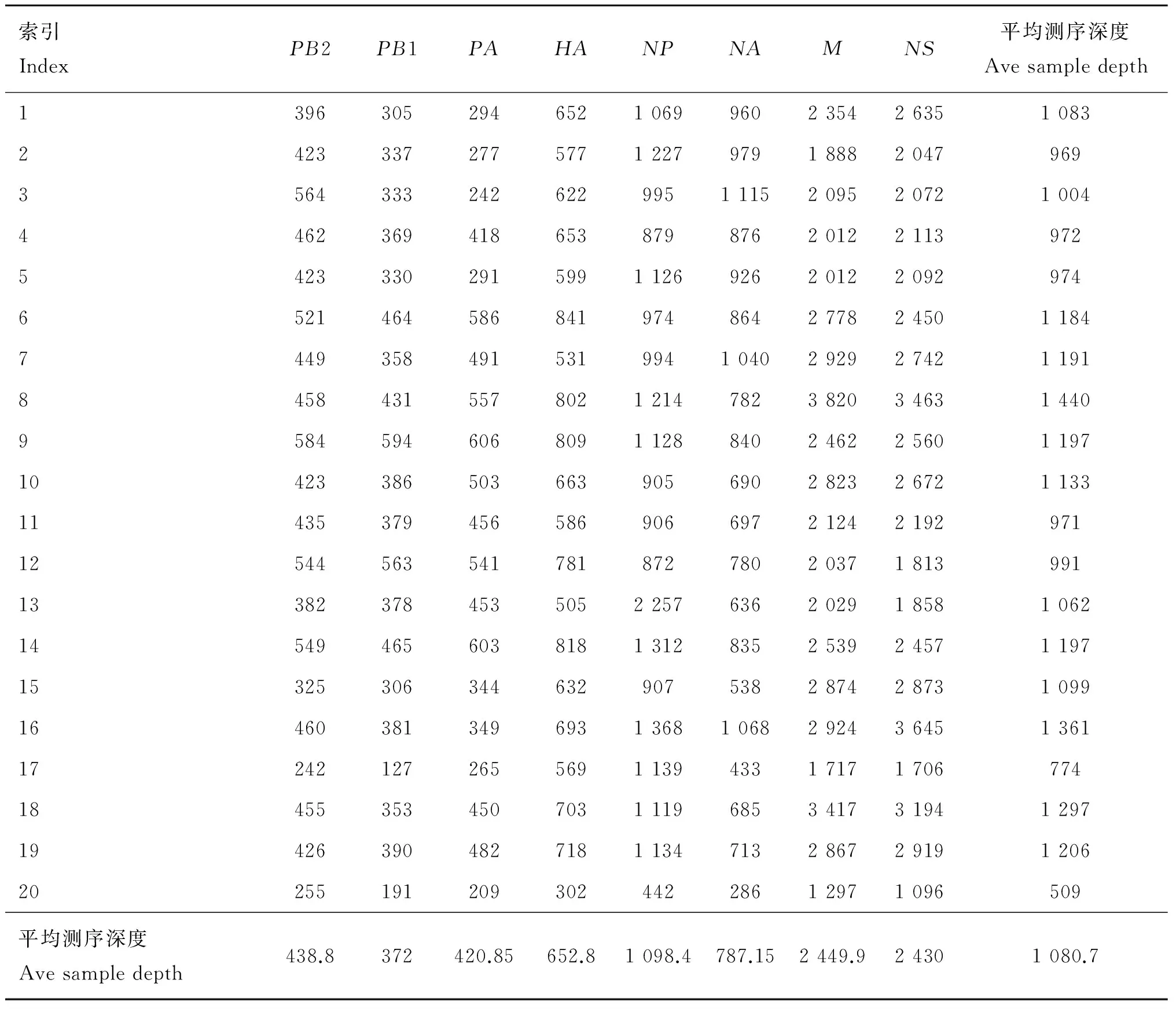
表2 毒株各节段的测序深度
2.4AIV全基因分子进化分析
20株分离株的8个节段的序列均登录GenBank,登录号:KT449572~KT449591(HA),KT449612~KT449631(NA),KT449592~KT449611(M),KT449632~KT449651(NP),KT449652~KT449671(NS),KT449672~KT449691(PA),KT449692~KT449711(PB1),KT449712~KT449731(PB2)。8个节段序列的分子进化分析结果见图1~3。
HA基因分析表明,此20株禽流感病毒均属于h9.4.2.5分支(代表株为A/chicken/Guangxi/55/2005),为我国近年来的优势流行分支。NA基因分析表明,此20株分离株均属于H9N2亚型毒株所在的分支。6个内部基因分子演化分析表明,作者所分离的20株毒株中有12株AIV病毒和H9N2亚型毒株亲缘关系更近;4株毒株的6个内部基因均和H7N9亚型毒株亲缘关系更近;2株毒株分别有4个基因和H7N9亚型毒株亲缘关系更近,1株毒株有4个基因和H10N8亚型毒株亲缘关系更近,1株毒株有2个基因和H10N8亚型毒株亲缘关系更近,其余内部基因的序列均与H9N2亚型病毒的亲缘关系较近。
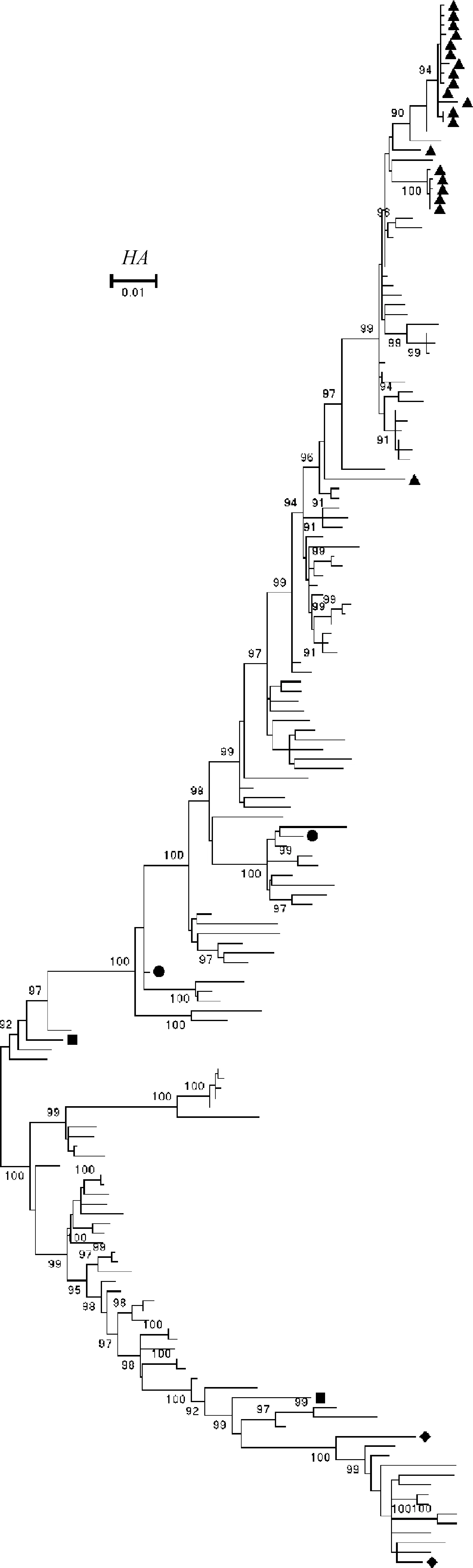
■.h9.4.2.4分支的代表毒株;●. h9.4.2.5分支的代表毒株;◆. h9.4.2.6分支的代表毒株;▲.所测20株分离株■. h9.4.2.4 clade representative virus;●. h9.4.2.5 clade representative virus;◆. h9.4.2.6 clade representative virus;▲. The 20 virus sequenced in the study图1 HA基因分子进化树Fig.1 Phylogenetic tree of HA gene
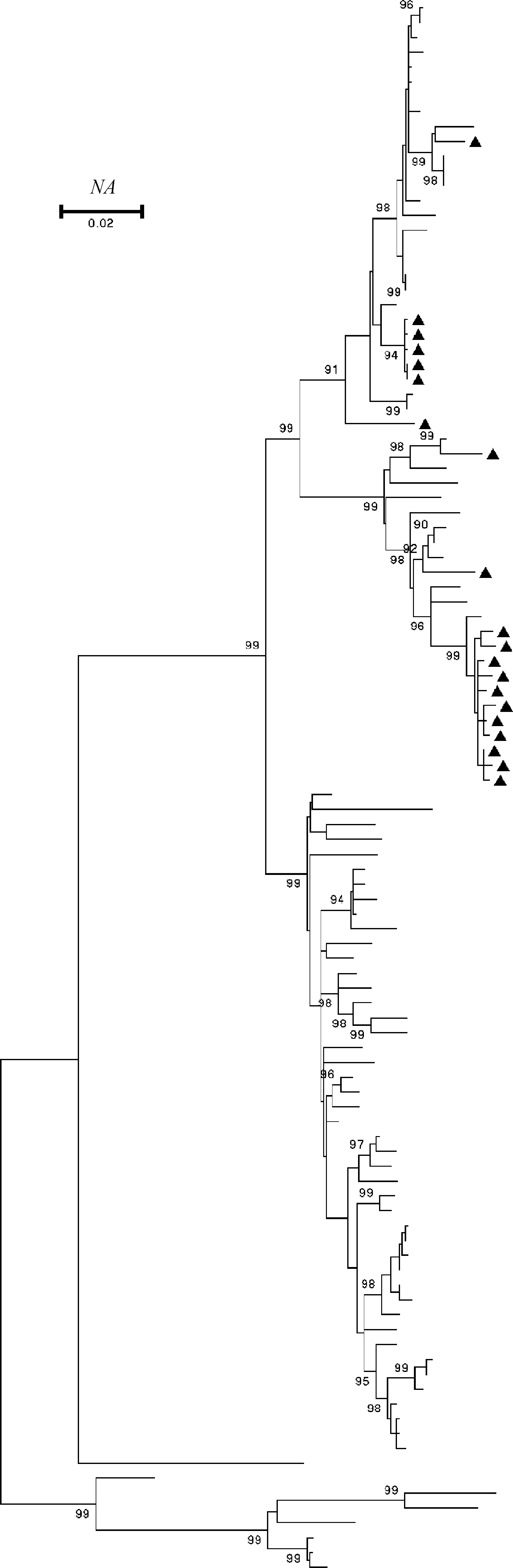
▲.所测20株分离株▲. The 20 virus sequenced in the study图2 NA基因分子进化树Fig.2 Phylogenetic tree of NA gene
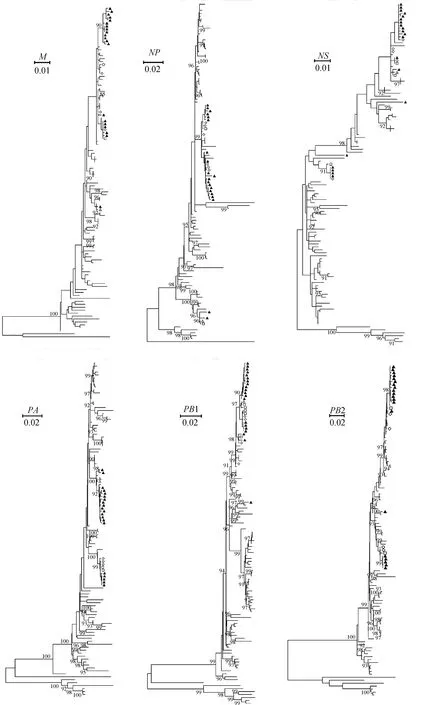
▲.所测20株分离株;◇.H7N9亚型禽流感病毒毒株;○.H9N9亚型禽流感病毒毒株;△.H10N8亚型禽流感病毒毒株;□.Mixed亚型禽流感病毒毒株▲. 20 virus sequenced in the study; ◇. H7N9 AIV;○. H9N9 AIV; △. H10N8 AIV; □. Mixed subtype AIV图3 M、NP、NS、PA、PB1和PB2基因分子进化树Fig.3 Phylogenetic tree of M, NP, NS, PA, PB1 and PB2 genes
3讨论
AIV是单股负链分节段的RNA病毒,其基因组复制时,具有RNA病毒特有的核酸碱基易变性,即抗原漂移(antigen drift)。不同的AIV同时感染同一细胞时,两个病毒的基因节段相互间还会发生基因重排,引起抗原转变(antigen shift)。所以,在自然界中每隔一定的时间,就会产生新的流感毒株引发流行。因而,测定AIV的全基因,掌握AIV的变异趋势,对于防控AIV尤为重要。
传统方法获取流感病毒全基因,是以每个节段的特异性引物分别扩增8个基因片段进行克隆,选择阳性克隆进行Sanger测序,然后进行序列拼接得到病毒基因组,该方法操作比较繁琐,既费时又费力。自高通量测序仪问世以来,高通量测序技术迅速发展,通量和读长都得到改善,具有标准化的文库构建流程,不需要常规实验中的细菌转化、培养以及单克隆挑选等,且短时间内可以获得大量有效的数据,能够快速获取病原体的遗传信息。Ion Torrent 高通量测序技术基于半导体技术,具有简单、快速、准确、灵活和低成本等显著优势,是中等规模测序项目的最佳选择,2012年A.S.Bowman等[15]利用Ion Torrent PGM高通量测序仪分析了H3N2亚型流感病毒的基因组。多项研究都对高通量测序用于禽流感病毒全基因的测序效果进行评价[16-17],证实该技术对AIV基因组的测序准确、快速,并能对AIV的遗传进化进行分析。
本文中作者采用Ion Torrent PGM高通量测序仪完成了20株AIV毒株的全基因测序,确定其均为H9亚型h9.4.2.5分支的低致病性毒株,NA基因均为N2亚型。结果显示,所采用的建库与测序方法能够测定流感病毒的全基因,并且测序深度较大,能满足序列分析的需要。
HA基因分子演化分析显示,所分离的20株毒株所在的h9.4.2.5分支,大部分均为我国2008年以后所分离的毒株。从毒株数量、时间和地理分布来看,在三级分支上,2007年以后第h9.4.2.5分支是中国H9亚型流感病毒的主要流行分支,这与尚飞雪等的结论一致[18]。本试验中有4株H9N2亚型毒株的6个基因均和H7N9亚型亲缘关系更近,2株H9N2亚型毒株分别有4个基因和H7N9亚型亲缘关系更近,推测某些H9N2亚型流感病毒的6个内部基因经过重配后,可能组成新的甲型H7N9流感病毒[19];还有1株H9N2亚型毒株的4个基因和H10N8亚型亲缘关系更近,1株H9N2亚型毒株的2个基因和H10N8亚型亲缘关系更近,与H.Chen等和H.Zhang等的观点一致,推测H9N2亚型流感病毒的某些内部基因经过重配后,可能会组成新的H10N8亚型流感病毒[20-21]。该方法的建立将进一步改善流感病毒基因组的测序方法,使该项工作的开展更为简便、快速、高效,更适用于分子流行病学调查中大量样品的测序分析,以及突发疫情的诊断与处置。
参考文献(References):
[1]FREIDL G S,BINGER T,MÜLLER M A,et al.Serological evidence of influenza A viruses in frugivorous bats from Africa [J].PLoSOne,2015,10(5):e0127035.
[2]TONG S,LI Y,RIVAILLER P,et al.A distinct lineage of influenza A virus from bats [J].ProcNatlAcadSciUSA,2012,109(11):4269-4274.
[3]TONG S,ZHU X,LI Y,et al.New world bats harbor diverse influenza A viruses [J].PLoSPathog,2013,9(10):e1003657.
[4]YOON S W,WEBBY R J,WEBSTER R G.Evolution and ecology of influenza A viruses [J].CurrTopMicrobiolImmunol,2014,385:359-375.
[5]HOFFMANN E,STECH J,GUAN Y,et al.Universal primer set for the full-length amplification of all influenza A viruses [J].ArchVirol,2001,146(12):2275-2289.
[6]GHEDIN E,SENGAMALAY N A,SHUMWAY M,et al.Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution [J].Nature,2005,437(7062):1162-1166.
[7]WANG M Z,TAI C Y,MENDEL D B.Mechanism by which mutations at his274 alter sensitivity of influenza a virus n1 neuraminidase to oseltamivir carboxylate and zanamivir [J].AntimicrobAgentsChemother,2002,46(12):3809-3816.
[8]ZHOU B,DONNELLY M E,SCHOLES D T,et al.Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses [J].JVirol,2009,83(19):10309-10313.
[9]DOMINGO E,BARANOWSKI E,RUIZ-JARABO C M,et al.Quasispecies structure and persistence of RNA viruses [J].EmergInfectDis,1998,4(4):521-527.
[10]CHEN R A,LAI H Z,LI L,et al.Genetic variation and phylogenetic analysis of hemagglutinin genes of H9 avian influenza viruses isolated in China during 2010-2012 [J].VetMicrobiol,2013,165(3-4):312-318.
[11]李建丽,陈恩林,李和平,等.H9N2亚型禽流感病毒NS1基因的进化分析[J].病毒学报,2008,24(3):220-226.
LI J L,CHEN E L,LI H P,et al.Genetic analysis of the nonstructural gene(NS1) of H9N2 avian influenza viruses isolated in China[J].ChineseJournalofVirology,2008,24(3):220-226.(in Chinese)
[12]EDGAR R C.MUSCLE:multiple sequence alignment with high accuracy and high throughput [J].NucleicAcidsRes,2004,32(5):1792-1797.
[13]LIU S,JI K,CHEN J M,et al.Panorama phylogenetic diversity and distribution of Type A influenza virus [J].PLoSOne,2009,4(3):e5022.
[14]WHO/OIE/FAO H5N1 Evolution Working Group.Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1) [J].EmergInfectDis,2008,14(7):e1.
[15]BOWMAN A S,SREEVATSAN S,KILLIAN M L,et al.Molecular evidence for interspecies transmission of H3N2pM/H3N2v influenza A viruses at an Ohio agricultural fair,July 2012 [J].EmergMicrobesInfect,2012,1(10):e33.
[16]BIDZHIEVA B,ZAGORODNYAYA T,KARAGIANNIS K,et al.Deep sequencing approach for genetic stability evaluation of influenza A viruses [J].JVirolMethods,2014,199:68-75.
[17]VAN DEN HOECKE S,VERHELST J,VUYLSTEKE M,et al.Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing [J].BMCGenomics,2015,16:79.
[18]尚飞雪,刘朔,蒋文明,等.近年来中国H9亚型禽流感分离株谱系分析[J].中国动物检疫,2012,29(4):51-53.
SHANG F X,LIU S,JIANG W M,et al.Phylogenetic analysis of H9 subtype avian influenza viruses isolated from China in recent years [J].ChinaAnimalHealthInspection,2012,29(4):51-53.(in Chinese)
[19]刘春艳,艾军红.甲型H7N9禽流感病毒的病毒学特征 [J].中国当代儿科杂志,2013,15(6):405-408.
LIU C Y,AI J H.Virological characteristics of avian influenza A H7N9 virus [J].ChineseJournalofContemporaryPediatrics,2013,15(6):405-408.(in Chinese)
[20]CHEN H,YUAN H,GAO R,et al.Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection:a descriptive study [J].Lancet,2014,383(9918):714-721.
[21]ZHANG H,XU B,CHEN Q,et al.Characterization of an H10N8 influenza virus isolated from Dongting lake wetland [J].VirolJ,2011,8:42.
(编辑白永平)
doi:10.11843/j.issn.0366-6964.2016.07.018
收稿日期:2015-10-30
基金项目:中国动物卫生与流行病学中心创新基金(2015IF-0004FF)
作者简介:王楷宬(1981-),女,山东青岛人,副研究员,主要从事动物病原学研究 *通信作者:王楷宬, E-mail:wangkaicheng@cahec.cn
中图分类号:S852.659.5
文献标志码:A
文章编号:0366-6964(2016)07-1443-08
Genome Analysis of the 20 Avian Influenza Virus Strains by High-Throughput Sequencing
WANG Kai-cheng*,ZHUANG Qing-ye,ZHANG Xiao-chun,QIU Yuan,WANG Tong,HOU Guang-yu,LIU Shuo,WANG Su-chun
(ChinaAnimalHealthandEpidemiologyCenter,Qingdao266032,China)
Abstract:To discuss the sequencing and analysis of avian influenza virus (AIV) genome by high-throughput sequencing,genomes of the 20 AIV strains isolated from eastern China were sequenced by Ion Torrent PGM.The advantage and disadvantage of the method were evaluated.The characteristic of genome and evolution were analyzed.The molecular evolution relationships of the other 6 internal genes were also analyzed.The results showed that the whole genomes of all the 20 strains were completely sequenced by Ion Torrent PGM.All the 20 viruses are restricted to h9.4.2.5 clade of H9 subtype low pathogenic strains.NA genes of all the viruses are N2 subtype.High-throughput sequencing can be used in the genome analysis of AIV strains.
Key words:high-throughput sequencing;avian influenza;genome;molecular evolution
