白酒微生物群落研究技术现状与二代测序数据分析方略
2016-08-03汪地强王和玉闫松显贵州茅台酒股份有限公司技术中心贵州仁怀564501
赵 亮,王 莉,汪地强,王和玉,闫松显(贵州茅台酒股份有限公司技术中心,贵州仁怀564501)
白酒微生物群落研究技术现状与二代测序数据分析方略
赵亮,王莉,汪地强,王和玉,闫松显
(贵州茅台酒股份有限公司技术中心,贵州仁怀564501)
摘要:白酒酿造实质上是微生物消长演替、代谢产物积累变化的过程,要搞清白酒本质,必须从微生物着手。微生物在酒醅发酵过程中以群体方式发挥作用,准确解析微生物群落构成及演替模式既是阐明白酒产量、质量的前提,也是进一步研究微生物功能及代谢的基础。针对各类微生物群落研究技术在白酒领域使用现状,对国内外研究报道做系统分析,发现纯培养以及不可培养DGGE/TGGE是目前研究白酒微生物群落的主流技术,而对微生物群落揭示较全面、系统的二代测序技术使用相对较冷,仅在外文期刊中报道相对较多。其次,通过应用现状成因分析,认为大多数研究者对测序数据的理解和分析水平不足是造成目前二代测序使用现状的主要原因。基于此,对测序后反馈的重要结果做系统解释,并提出5类分析微生物群落的重要多元统计方法以及各类方法分析策略与使用方案,旨在为白酒微生物群落研究者提供可行性高、参考性强的二代测序数据分析方略。
关键词:白酒; 二代测序; 微生物多样性; 多元统计方法; 酿酒微生物
白酒作为我国传统发酵食品,历史悠久,长期受到人民大众的喜爱。中国传统白酒,是以富含淀粉质的粮谷类为原料,并辅以大曲作糖化发酵剂,采用固态、半固态或液态发酵技术,经蒸馏、贮存以及勾调过程酿制而成的含酒精饮料[1],被列为世界著名六大蒸馏酒之一。结合白酒生产工艺可知,白酒酿造过程实质上是相关微生物经历消长演替,代谢产物变化积累的过程。这些相关微生物统称白酒微生物,既包括自然接种的天然微生物,也包括人工选育的纯种微生物;既包括糖化菌、发酵菌等有益微生物,也包括导致苦味和酸败的有害菌[2]。
要解析白酒本质,必须从白酒微生物研究入手。微生物在白酒发酵中是以群体的方式发挥作用,因而准确解析白酒微生物群落既是阐明白酒产量与质量的前提,也是进一步研究微生物功能及代谢变化的重要基础。微生物群落研究,亦即微生物生态研究,主要揭示微生物在被研究环境(环境在这里特指微生物群落的栖息场所,如土壤、酒醅、大曲、水体、动植物组织等)内的多样性,以及微生物与环境、微生物之间相互作用关系,微生物如何维持在环境中特有的多样性水平。据此,微生物生态研究目前大致分为两个领域:①微生物多样性领域,包括对环境内各微生物组分进行系统分类鉴定、定量,以及对微生物群落构成多样化进行衡量;②微生物活性领域,包括微生物区系(适应环境,并可在环境中正常生长繁殖的主要微生物组成的群落结构)研究,对环境理化特征产生影响或维持环境内特定微生物多样性水平的主要群落结构研究,同时包括微生物与微生物、微生物与环境间生态响应关系的研究[3]。
微生物群落研究至今,其遗传信息获取技术大体包括[4-6]:①传统微生物纯培养法;②以PCR为基础的微生物指纹图谱法:主要包括DGGE/TGGE、AFLP、SSCP、RFLP、RAPD、Q-PCR、T-RFLP;③核酸探针杂交技术:主要包括FISH、生物芯片;④磷脂脂肪酸法(PLFA);⑤Biolog微平板法;⑥宏基因组测序法。白酒微生物群落的信息获取与分析,同样依赖上述6类技术手段。本文将进行以下研究:(1)通过文献搜集统计,分析白酒微生物群落研究技术现状;(2)针对近年来逐渐成为微生物群落研究主流的前沿技术——第二代测序技术(又称宏基因组或高通量测序技术),剖析其在白酒微生物生态领域的应用现状及前景;(3)针对第二代测序技术的应用,提出研究者如何通过商业测序公司返回的测序数据分析结果,合理利用、解释自己的研究内容,并根据国际上分析微生物生态数据较为主流的统计方法,针对白酒微生物群落高通量数据的深度挖掘给予相关建议。
1 白酒微生物群落研究技术现状
1.1文献统计
为尽可能全面获得白酒微生物群落研究信息,笔者分别以“白酒”“大曲”“酒醅”为搜索关键词,结果如图1所示,从中国学术期刊全文数据库(CNKI)筛查到与微生物群落研究内容相关的文献报道58篇,分别出自13类中文学术期刊,其中《酿酒科技》文章刊登量最多,占筛查文献总数的50%(29/58)。同样,笔者以Chinese liquor、Daqu、Fermented grains为搜索关键词,通过SCI科学引文索引数据库(Web of Science)筛查到85篇相关文献,分别出自20类外文学术期刊,其中J I Brewing和World J Microb Biot文献产出量最高,分别占筛查文献总数的16.5%(14/85)和12.9%(11/85)。根据中文、外文筛查文献总数显示,白酒微生物群落研究目前正处于发展阶段,文献报道量不高,且期刊分布类型较少。此外,外文文献多数发表于影响力(Impact factor)偏低的期刊,微生物学科权威性期刊,以及高影响力综合性期刊几乎没有白酒微生物群落研究的报道。根据我们对环境微生物领域发表文章的水平评估发现,期刊影响力越高,文章中涉及的微生物信息获取方法越先进,并且后期生物信息分析深度、严密,所得结论可靠性强。
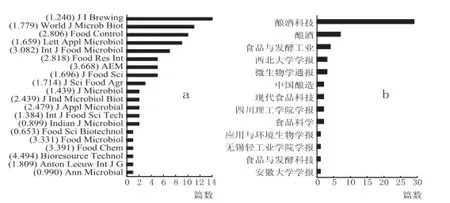
图1 白酒微生物群落研究文章发表统计
1.2白酒微生物群落研究技术概况
为直观反映微生物群落研究技术在白酒领域应用现状,我们对各类技术方法发表于图1中各期刊的频率做汇总,并以期刊名与技术方法作响应变量,发表频率作解释变量,进行对应分析(Correspondence analysis,CA)并生成双标图(图2)。图2中各圆代表不同研究技术,圆面积表示此技术使用频率,面积越大,使用频率越高,反之越低;圆与期刊名在图中距离越近,表示发表于该期刊文章使用此技术的相对频率越高;圆越靠近坐标原点,表明此技术越普及。由图2可知,国内文献多采用纯培养、DGGE、以及Biolog技术分析白酒微生物群落,特别是纯培养技术的应用在国内文献的占比与普及程度最高。相对于此,其他一些技术如PLFA和PCR-SSCP,分别与中国酿造、四川理工学院学报在图中距离最近,说明两类技术仅分别在这两种期刊文献中较常使用,在其他期刊文献中很少或不被使用。国外SCI文献中,DEEG技术使用频率和普及程度最高,二代测序技术普及程度次之,但使用频率略低于Q-PCR与纯培养技术。通过整体对照发现,纯培养技术无论在国内或国际文献中使用频率均高,这与微生物在白酒酿造中实际应用有关。以DGGE、Q-PCR、二代测序和克隆文库测序为代表的非培养技术,DGGE无论使用率和普及程度在国内外文献中均占一席之地,这可能与DGGE技术使用成本低廉、群落中不可培养优势微生物分离效果较好有关。然而,从微生物群落研究现阶段发展来看,二代测序技术无疑是揭示环境微生物群落成分及结构最理想手段,但此技术仅在国际期刊文献中有一定程度普及,且使用频率并非最高。针对此现象,我们认为潜在原因有两点:(1)二代测序技术成本偏高,市场单价随测序样本量大小以及测序公司不同基本在每样400~700元之间浮动,此外,一项研究需要较大样本量才能满足此类研究对数据统计的严格需求,以致总体研究成本偏高;(2)实验室经费充足,但对二代测序技术了解不足,面对测序公司返回结果无力适从,多数情况仅能以测序公司返回的初级结果支撑文章论点,导致研究成果无法在权威性强、高影响力的杂志发表。本文后续内容将针对潜在原因2,深入探讨二代测序反馈结果中可能涉及到的几类重要多样性指数意义、以及如何借助多元统计分析对反馈数据做进阶挖掘,以获得准确度高、生物学意义充实的研究结论。

图2 对应分析双标图展示不同研究技术在各期刊文献使用频度
2 二代测序数据分析策略
2.1测序平台与反馈数据简介
目前二代测序的主要平台代表有Illumina公司的Solexa基因组分析仪(Illumina Genome Analyzer)、罗氏公司(Roche)的454测序仪(Roch GS FLX sequencer)和ABI的SOLiD测序仪(ABI SOLiD sequencer)。微生物多样性分析中,以Illumina及454测序平台应用最为广泛。各平台测序原理已有报道做系统探讨[7-8],在此不加赘述。测序公司对微生物总DNA样品完成可变区PCR扩增—建库—上机测序—数据收集及质控—数据分析等一系列工作后,将结果返给客户。因测序公司不同,返回结果在形式及细节上存在较大区别,但核心内容无外乎4个方面:①序列数据:包含命名为“rawdata”和“cleandata”2个文件夹,前者存放下机后原始序列文本,后者存放原始序列经质量控制后可用序列文本,后续分析均基于“cleandata”文件内容完成;②OTU数据:主要包含两类信息,其一是以样本名为列变量,OTU名(OTU通常命名为Denovoi,i=1,2,3,...,OTU总数n)为行变量的n×p矩阵,矩阵变量为每个OTU在各样本中序列数(reads),矩阵第p列为各行OTU对应从界到属系统分类信息(能鉴定到种的会延伸至种);其二是每个OTU代表序列(以序列长度最长,质量最好的序列作为代表序列),通常以fasta格式存放;③多样性指数:包括α、β两种多样性指数,通常以excel表格文件存放;④相对丰度分布表:通常给出从界到属(L1—L6,能鉴定到种的延伸至种L7)各分类水平下相对丰度表,每个分类水平对应一个相对丰度表。除此之外,不同测序公司会基于上述结果②—④为客户免费提供某些进阶分析服务,如组间差异性分析,绘制系统进化树、相对丰度热图、相对丰度柱状堆积图、面积图、样本或组间共享OTU数目的Venn图等。
2.2多样性指数
经查阅众多相关文献发现,针对多样性指数的生物学解释及剖析通常作为首要内容位于文段前部,已成为构架文章主体不可或缺的内容。另外,文章重要结论的产出和后续内容的延伸均需要此部分内容做支撑。因而,笔者需要对多样性指数做一重点讨论。
各测序公司在返给客户的结果中会提供α、β两类多样性指数[8],α多样性指数原意为生境内多样性指数,此处可理解为样品OTU组成多样性,如果样品有分组,组内各样品将整合计算,得出组内OTU组成多样性。β多样性指数原意为沿环境梯度不同生境群落之间物种组成的相异性或物种沿环境梯度的更替速率,此处可理解为样品间或组间OTU组成差异度。通常测序公司会利用Mothur[9]http://www.mothur.org/或Qiime[10]http://qiime.org/软件计算出Shannon-Wiener、Simpson、Chao1、ACE和PD 5种指数衡量α多样性。Shannon-Wiener指数借助信息论原理,用于衡量样本或组内下一条采集序列OTU归属不确定程度,不确定程度越高,多样性水平越高。Simpson指数表示样本或组内随机抽取2条序列属于不同OTU的概率,概率越大多样性水平越高;Chao1指数是利用仅包含1条和2条序列的OTU数、结合观察到的OTU数,估计样本或组内OTU总数;ACE与Chao1类似,也是用于估计样本或组内OTU总数的指数,但算法与Chao1不同,ACE主要借助稀疏OTU数结合观察到的OTU数估计OTU总数。一般默认序列数小于或等于10 的OTU为稀疏OTU,用户也可根据分析要求自己定义稀疏OTU序列数阈值。PD指数的“PD”取自“Phylogenetic diversity”两个单词首字母,意指系统进化多样性指数。它在估计样本或组内多样性水平时,重点衡量不同OTU间整体亲缘度,PD越高,OTU间整体亲缘度越远。β多样性主要利用Bray-Curits和UniFrac两类距离矩阵[11-12]衡量,距离越大说明多样性越高。Bray-Curits距离主要衡量样本间或组间OTU成分差异度,距离越大,差异度越高;UniFrac距离主要衡量样本间或组间OTU平均亲缘度,距离越大,亲缘度越远。同时,两类矩阵又分加权(Weighted)和不加权(Unweighted)两种,具体差异见表1。
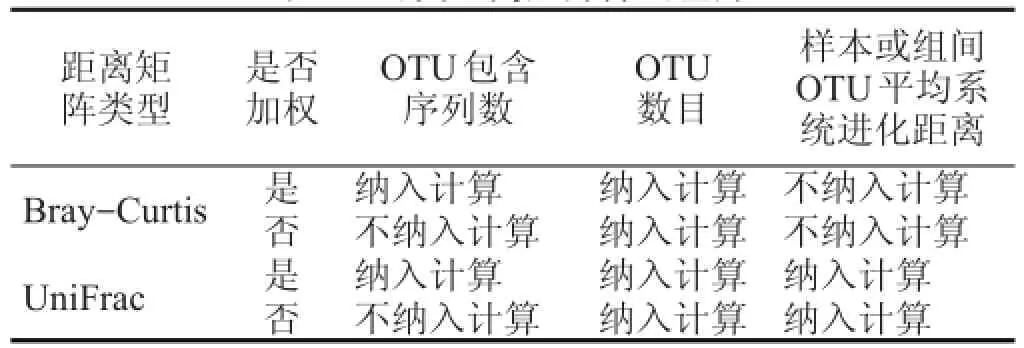
表1 两类距离矩阵算法差异
2.3数据多元统计方法
使用多元统计处理测序后数据,旨在挖掘群落间隐藏或预判的相互关系和组成模式,以及与这种关系或模式存在因果联系的影响因素。以R[13]https://www.r-project.org/或Canoco[14]软件分析为例,需要以样本名作行变量,即每一个样本代表一个群落,物种名作列变量构建一套群落分析表,表中数值为物种序列读数或相对丰度,形式如表2。物种在这里泛指OTU或各种分类名,如在纲水平解析群落,物种名即为不同纲名。若假设分析群落存在一定分布模式,或群落分布受某些环境梯度影响,需要再构建一套用于解释群落的环境变量表,此表行变量为样本名,且在表中顺序必须与群落分析表中行变量顺序一致,列变量为分组名及其他环境变量名,制表形式如表4。这两套表格的构建可完成后续如聚类、排序、差异性、判别、回归及相关性5大类分析。如表4内容所示,这些分析可帮助我们全面理解酿酒微生物群落构成、演替、以及与环境间相互作用规律。下文将针对表4内容,对5大类分析的使用作逐一讨论。

表2 群落分析表表格样式

表3 环境变量表表格样式
2.3.1聚类分析
聚类分析是利用列变量在行变量间变异程度,主要以树图形式表示行变量间相似或非相似度水平。如此,越相似的行变量,在图形中越被聚拢靠近;反之,则相互远离。在这种趋势推动下,行变量将在一定尺度被聚集成多个组群。如表4所示,聚类分析方法众多,但分析过程与分析结果的解读方式基本一致(K-mean clustering除外)。以微生物群落研究最常用的聚类法UPGMA为例,Bokulich等将属于Chardonnay musts的数据集抽离出来,构成如表2形式数据集,并结合每个OTU代表序列,构建样本间weighted UniFrac距离,基于此距离利用UPGMA法对样本聚类,生成聚类图Fig.1A,后经探索分析,发现聚群模式主要由样本采集区域不同而产生。样本在聚类树图中的聚群方式和类群数目会随研究者定义的相似度或非相似度尺度的不同而异。至于样本在何种尺度聚群,研究者需要分析探索,通常聚群方式是否最佳,要看此方式是否能得到最有力的生物学解释。如Bokulich等[61]在Fig.1A中确定的聚群方式可被采样区域的不同所解释。聚类分析是针对样本组群分析中运算最简单的方法,其缺陷在于研究者无法从中直接获得样本最佳组群数及聚群模式,因而在微生物群落分析中仅充当探索性工具使用。
2.3.2判别分析
研究者在分析群落数据时,通常认为样本可能会因群落成分差异而划分成多个组群,而这个组群的形成必然与实验设计有关。如取自窖内的酒醅样本分别来源于3个不同发酵时期,研究者会设想属于同一发酵时期的样本具有较高微生物群落相似度,以致样本会形成3个组群,分别对应不同发酵时期。判别分析的作用在于评测这种假设群的准确度;同时假设群一旦成立,分析生成的判别模型可对新样本做判断归类,另外可为研究者找出推动各样本被归于各假设群的重要变量。实际分析时,群落分析表2作解释变量,表3任一列分组模式,即假设组作响应变量。表4罗列出5种常规分析使用时各有侧重,DAPC和CAPDA侧重分析假设组准确性,CAPDA可专门针对距离或相似度矩阵做判别分析,若行变量(样本数)过多,计算时间相对较长。Rf同时具备分析假设组准确性及找出相对重要物种的作用,且特别适合大数据集分析。但要得到准确率较高的结果,研究者需要对此方法具备一定实用经验。LDA effect size和CDA侧重寻找相对重要物种,两类分析适合在样本量适中或较小的情况下使用。实际分析时,需要根据自己的研究目的及数据特征,从中选取几种方法配合使用。

表4 5大类多元统计分析内容描述
2.3.3排序分析
排序分析是微生物群落研究主流分析方法。样本间或物种间关系可能在PCA、CA、NMDS、PCoA、DCA分析中得到一定展示,但促使这种关系形成的环境梯度不在此排序分析中直接体现,需要利用样本或物种的重要排序轴(通常为前2个排序轴)得分与相应的环境变量建立联系,方可表现物种或样本分布受环境梯度的影响趋势,因而此类排序也被称为间接梯度排序。与此相对,物种或样本分布与环境梯度的关系可在RDA、db-RDA、CCA、DCCA分析中被直接体现,故此类排序又被称为直接梯度排序。两类排序具体算法本文不做赘述,研究者可通过表4及图3了解两类排序法的区别及使用策略。几种直接梯度排序法分析目的及解释途径基本相同,至于选取哪种排序法最佳,可利用DCA分析对表2排序。DCA第一梯度轴长小于3,建议使用线性模型排序法RDA或db-RDA;轴长大于4建议使用单峰模型排序法CCA或DCCA;轴长介于3-4,线性模型或单峰模型排序法均可使用。

续表4 5大类多元统计分析内容描述

图3 排序法使用关系流程
2.3.4相关性及回归分析
两类分析既有联系又有区别。解决两个一元数值型变量间直线相关性问题,可使用相关性分析,Kendall和Spearman秩相关性分析可针对解决数值分布非正态分布或分布模式未知的变量间相关性问题。一旦使用相关性分析发现2个变量间存在高度相关,建议使用线性回归进一步探索一个变量随另一个变量的变化趋势。相关性分析中2个变量不存在因果关系,即无解释变量和响应变量之分。分析结果中的R(R∈[-1,1])仅作为衡量2个变量间相关性程度及方向的参数,即-1≤R<0时,2个变量间呈负相关,越接近-1,负相关程度越大;0<R≤1时,2个变量间呈正相关,越接近1,正相关程度越大;当R=0时,2个变量间无相关性。相关性分析不能将R值当作回归分析中的自变量系数,用于1个变量的数值变化推测另一个变量的数值变化。回归分析相比相关性分析更复杂,变量数值分布模式需要提前预知。但可解决变量间直线或曲线相关性问题。用于回归的变量必须确定自变量与应变量,两类变量均可以是一元或多元变量。若应变量为多元变量,建议使用PLS作回归分析。在做微生物群落分析时,需根据研究者重点考察的问题对象选择相应分析方案,并建议在具体研究时,两类分析方法协同并用。
2.3.5差异性分析
顾名思义,此分析用于检验属于不同组的数量间是否有差异,以及衡量差异程度有多大。对于一元变量差异性分析,可借助均值或中位数比较各组数量占有高低。例如同一物种的相对丰度或序列数在不同组间差异性的比较属于一元变量差异性分析,分析方法通常使用Anova或npAnova。但笔者发现,多数微生物群落研究者也许统计知识不足,数据列在不符合正态分布及方差齐性的情况下仍强制套用参数检验Anova作分析,导致结果很可能出现偏差。微生物群落研究中,所获数据在做具体分析情形下,原始数列直接符合正态分布的情况很少,通常需要对原始数列做对数或标准化转换,方可使用基于正态分布的统计方法。然而,多数情形下,数据即使做了转换,仍旧不符合正态分布。非参数检验法不用考虑数据列的分布限制,适用范围广,因而此类方法也是被大量国际期刊频繁使用、报道的原因。笔者建议,在处理庞杂的微生物群落数据时,组间差异性分析优先使用非参数检验法,然后针对需要重点考察或与预想偏差较大的结果,再次使用参数检验法。如果数列符合参数检验规则,分析结果以参数检验为准,这是因为非参数检验虽条件宽松,但包含信息量小,功效相比参数检验略逊色。至于多元变量组间差异性分析,目前微生物群落研究领域主要采用非参数分析法,如表4所示(Manova除外)。此处需要说明,基于限制排序法的组间差异性分析,可通过对干扰变量的限制隔离出目标分组所产的净效应。例如,表2群落来自于不同发酵时期窖内酒醅样品,并且样品取自不同窖池、窖内不同深度。每个样品还测得酒精含量、酸度、温度3种理化参数。将样本理化特征以及采样信息录入表3,则有3种分组模式(发酵时期、窖内深度、窖池区别),3种数值型环境变量(酒精含量、酸度、温度),总共6个环境因子。现需要求得发酵时期是否对微生物群落成分有显著性差异,且发酵时期单独产生的差异有多大,则可以在分析时以发酵时期作解释变量,另外5种因素作控制变量,以此排除控制变量对微生物群落产生的额外影响,从而获得发酵时期的净效应。
参考文献:
[1]余乾伟.传统白酒酿造技术[M].北京:中国轻工业出版社,2010.
[2]王旭亮,王德良,韩兴林,等.白酒微生物研究与应用现状[J].酿酒科技,2009(6):88-91.
[3]Xu J P.Microbial ecology in the age of genomics and metagenomics:concepts,tools,and recent advances[J]. Molecular Ecology,2006,15:1713-1731.
[4]冯佩英,陆春,朱国兴.DGGE/TGGE技术在微生物基因分类鉴定中的应用[J].国外临床医学生物化学与检测学分册,2005 (26):95-97.
[5]王晓丹,谢晓丽,胡宝东,等.现代微生物分类鉴定技术在白酒酿造中的应用[J].中国酿造,2015(34):5-9.
[6]Smith C J,OsbornAM.Advantages and limitations of quantitative PCR(Q-PCR)-based approaches in microbial ecology[J].Fems Microbiology Ecology,2009,67:6-20.
[7]杜玲,刘刚,陆健,等.高通量测序技术的发展及其在生命科学中的应用[J].中国畜牧兽医,2014(41):109-115.
[8]张金屯.数量生态学[M].北京:科学出版社,2004.
[9]Schloss P D,Westcott S L,Ryabin T,et al.Introducing mothur: open-source,platform-independent,community-supported software for describing and comparing microbial communities [J].Applied Environmental Microbiology,2009,75(23):7537-7541.
[10]Caporaso J G,Kuczynski J,Stombaugh J,et al.QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods,2010,7:335-336.
[11]Lozupone CA,Knight R.UniFrac:Anew phylogenetic method for comparing microbial communities[J].Applied Environmental Microbiology,2005,71(12):8228-8235.
[12]Faith D P,Minchin P R,Belbin L.Compositional dissimilarity as a robust measure of ecological distance[J].Vegetatio,1987,69,57-68.
[13]R Development Core Team.R:ALanguage and Environment for Statistical Computing[M].Vienna:the R Foundation for Statistical Computing,2015.
[14]Ter Braak C J F,Smilauer P.CANOCO Reference Manual and User's Guide to Canoco for Windows:Software for Canonical Community Ordination[R].Ithaca:Microcomputer Power,1998.
[15]Hartigan JA,Wong MA.AK-means clustering algorithm[J]. Applied Statistics,1979,28,100-108.
[16]Kaufman L,Rousseeuw P J.Finding Groups in Data:An Introduction to ClusterAnalysis[M].New York:Wiley,1990.
[17]Sneath P HA,Sokal R R.Numerical Taxonomy[M].San Francisco:Freeman,1973.
[18]Saitou N,Nei M.The neighbor-joining method:a new method for reconstructing phylogenetic trees[J].Molecular Biology and Evolution,1987,4:406-425.
[19]Segata N,Izard J,Waldron L,et al.Metagenomic biomarker discovery and explanation[J].Genome Biology,2011,12(6):R60.
[20]Jombart T,Devillard S,Balloux F.Discriminant analysis of principal components:a new method for the analysis of genetically structured populations[J].BMC Genetics,2010,11:94.
[21]LiawA,Weiner M.Classification and regression by Random Forest[J].R News,2002,2(3):18-22.
[22]Bartlett M S.Further aspects of the theory of multiple regression[J].Proceedings of the Cambridge Philosophical Society,1938,34:33-34.
[23]Cooley W W,Lohnes P R.Multivariate DataAnalysis[M]. New York:Wiley,1971.
[24]Gittins R.CanonicalAnalysis:AReview withApplications in Ecology[M].Berlin:Springer,1985.
[25]Husson F,Le S,Pages J.Exploratory MultivariateAnalysis by Example Using R[M].CRC Press,2010.
[26]Anderson M J,Willis T J.Canonical analysis of principal coordinates:a useful method of constrained ordination for ecology[J].Ecology,2003,84:511-525.
[27]Legendre P,Anderson M J.Distance-based redundancy analysis:testing multispecies responses in multifactorial ecological experiments[J].Ecological Monographs,1999,69:1-24.
[28]Benzecri J P.CorrespondenceAnalysis Handbook[M].New York:Dekker,1992.
[29]Mardia K V,Kent J T,Bibby J M.MultivariateAnalysis[M]. London:Academic Press,1979.
[30]Cailliez F.The analytical solution of the additive constant problem[J].Psychometrika,1983,48:305-308.
[31]Gower J C.Some distance properties of latent root and vector methods used in multivariate analysis[J].Biometrika,1966,53:325-338.
[32]Gower J C,Legendre P.Metric and Euclidean properties of dissimilarity coefficients[J].Journal of Classification,1986,3:5-48.
[33]Legendre P,Gallagher E D.Ecologically meaningful transformations for ordination of species data[J].Oecologia,2001,129:271-280.
[34]Legendre P,Legendre L.Numerical Ecology[M].2nd ed. Amsterdam:Elsevier Science BV,1998.
[35]Lingoes J C.Some boundary conditions for a monotone analysis of symmetric matrices[J].Psychometrika,1971,36:195-203.
[36]Minchin P R.An evaluation of relative robustness of techniques for ecological ordinations[J].Vegetatio,1987,69:89-107.
[37]Hill M O,Gauch H G.Detrended correspondence analysis:an improved ordination technique[J].Vegetatio,1980,42:47-58.
[38]Oksanen J,Minchin P R.Instability of ordination results under changes in input data order:explanations and remedies [J].Journal of Vegetation Science,1997,8:447-454.
[39]Legendre P,Legendre L.Numerical Ecology[M].3rd ed.Elsevier,2012.
[40]Ter Braak C J F.Canonical CorrespondenceAnalysis:a new eigenvector technique for multivariate direct gradient analysis [J].Ecology,1986,67:1167-1179.
[41]Chambers J M.Linear models[M]//Chambers J M,Hastie T J. Statistical Models in S.Chapman and Hall,1991.
[42]Wilkinson G N,Rogers C E.Symbolic descriptions of factorial models for analysis of variance[J].Applied Statistics,1973,22:392-399.
[43]DobsonAJ.An Introduction to Generalized Linear Models [M].London:Chapman and Hall,1990.
[44]Hastie T J,Pregibon D.Generalized linear models[M]// Chambers J M,Hastie T J.Statistical Models in S.Chapman and Hall,1991.
[45]McCullagh P,Nelder JA.Generalized Linear Models[M]. London:Chapman and Hall,1989.
[46]Venables W N,Ripley B D.ModernApplied Statistics with S [M].New York:Springer,2002.
[47]Laird N M,Ware J H.Random-effects models for longitudinal data[J].Biometrics,1982,38:963-974.
[48]Lindstrom M J,Bates D M.Newton-Raphson and EM algorithms for linear mixed-effects models for repeatedmeasures data[J].Journal of theAmerican Statistical Association,1988,83:1014-1022.
[49]Geladi P,Kowlaski B.Partial least squares regression: a tutorial[J].Analytica ChimicaActa,1986,185:1-17.
[50]Hoskuldsson,A.PLS Regression Methods[J].Journal of Chemometrics,1988,2:211-228.
[51]Bates D M,Watts D G.Nonlinear RegressionAnalysis and Its Applications[M].Wiley,1988.
[52]Bates D M,Chambers J M.Nonlinear models[M]//Chambers J M,Hastie T J.Statistical Models in S.Chapman and Hall,1991.
[53]Best D J,Roberts D E.AlgorithmAS 89:the upper tail probabilities of spearman's rho[J].Applied Statistics,1975,24:377-379.
[54]Hollander M,Wolfe DA.Nonparametric Statistical Methods [M].New York:John Wiley&Sons,1973:185-194.
[55]Gittins R.CanonicalAnalysis:AReview withApplications in Ecology[M].Berlin:Springer,1985.
[56]Clarke K R.Non-parametric multivariate analysis of changes in community structure[J].Australian Journal of Ecology,1993,18:117-143.
[57]Hand D J,Taylor C C.MultivariateAnalysis of Variance and Repeated Measures[M].Chapman and Hall,1987.
[58]Mielke PW,Berry K J.Permutation Methods:ADistance FunctionApproach[M].New York:Springer,2001.
[59]Anderson M J.Anew method for non-parametric multivariate analysis of variance[J].Austral Ecology,2001,26:32-46.
[60]Anderson M J.Distance-based tests for homogeneity of multivariate dispersions[J].Biometrics,2006,62:245-253.
[61]Bokulicha NA,Thorngated J H,Richardson P M,et al. Microbial biogeography of wine grapes is conditioned by cultivar,vintage,and climate[J].Proceedings of the National Academy of Sciences of the United States ofAmerica,2013,25:E139-E148.
优先数字出版时间:2016-04-01;地址:http://www.cnki.net/kcms/detail/52.1051.TS.20160401.1330.003.html。
中图分类号:TS262.3;TS261.1;Q93-3
文献标识码:A
文章编号:1001-9286(2016)07-0088-09
DOI:10.13746/j.njkj.2016048
收稿日期:2016-02-18
作者简介:赵亮(1983-),男,博士,从事环境微生物多样性研究,已发表论文数篇,E-mail:20064827@qq.com。
Present Status in Research Technology of Liquor-making Microbial Communities&Next-generation Sequencing Data Analysis
ZHAO Liang,WANG Li,WANG Diqiang,WANG Heyu and YAN Songxian
(Technical Center of Maotai Distillery Co.Ltd.,Renhuai,Guizhou 564501,China)
Abstract:Liquor-making is essentially the process of microbial communities succession and microbial metabolites accumulation and transformation.To reveal the mystery of liquor,we should start from microbial research.Microbes in communities are,in general,advancing the fermentation of grains.The analysis of the composition and the succession of microbes is not only the premise to evaluate the yield and the quality of liquor,but also the basis for exploring the functions and metabolites of microbes.In this paper,aiming at the current application status of microbial research technology in liquor-making field,we found that culture-based approach and culture-independent DGGE/TGGE techniques are mainly applied in microbial community research nowadays,while next-generation sequencing technique is rarely adopted(reported only in foreign journals).Researchers not capable of understanding or manipulating the sequencing data might be the main reason for the poor use of nextgeneration sequencing technique.In view of such problem,systematical and straightforward interpretation of sequencing data were made in this review.Furthermore,five categories of multivariate analysis which could deeply and comprehensively present the microbial information were recommended,and we also provided the strategies and usages of these analytic approaches for reference.
Key words:Baijiu;next-generation sequencing;microbial diversity;multivariate statistical methods;liquor-making microbes
