Zika virus spreading in South America: evolutionary analysis of emerging neutralizing resistant Phe279Ser strains
2016-07-25MartaGiovanettiTeresaMilanoLuizCarlosAlcantaraLauraCarcangiuEleonoraCellaAlessiaLaiAlessandraLoPrestiStefanoPascarellaGianguglielmoZehenderSilviaAngelettiMassimoCiccozziDepartmentofInfectiousParasiticandImmunomediatedDiseases
Marta Giovanetti*, Teresa Milano, Luiz Carlos Alcantara, Laura Carcangiu, Eleonora Cella,, Alessia Lai, Alessandra Lo Presti, Stefano Pascarella, Gianguglielmo Zehender, Silvia Angeletti, Massimo Ciccozzi,Department of Infectious Parasitic and Immunomediated Diseases, National Institute of Health, Rome, ItalyDepartment of Biology, University of Rome ‘Tor Vergata’, Rome, ItalyLaboratory of Hematology, Genetic and Computational Biology, Gonçalo Moniz Research Center, Oswaldo Cruz Foundation (LHGB/CPqGM/ FIOCRUZ), Salvador, Bahia, BrazilDipartimento di Scienze Biochimiche ‘A. Rossi Fanelli’, Università La Sapienza, 00 Roma, ItalyDepartment of Public Health and Infectious Diseases, Sapienza University of Rome, Rome, ItalyLaboratory of Infectious Diseases and Tropical Medicine, University of MilanClinical Pathology and Microbiology Laboratory, University Hospital Campus Bio-Medico of Rome, Rome, ItalyUniversity Campus Bio-Medico, Rome, Italy
ABSTRACT
Objective: To investigate the genetic diversity of Zika virus (ZIKV) and the relationships existing among these circulating viruses worldwide. To evaluate the genetic polymorphisms harboured from ZIKV that can have an infl uence on the virus circulation. Methods: Three diff erent ZIKV dataset were built. The fi rst dataset included 63 E gene sequences, the second one 22 NS3 sequences and the third dataset was composed of 108 NS5 gene sequences. Phylogenetic and selective pressure analysis was performed. The edited nucleic acid alignment from the Envelope dataset was used to generate a conceptual translation to the corresponding peptide sequences through UGene software. Results: The phylogeographic reconstruction was able to discriminate unambiguously that the Brazilian strains are belonged to the Asian lineage. The structural analysis reveals instead the presence of the Ser residue in the Brazilian sequences (however already observed in other previously reported ZIKV infections) that could suggest the presence of a neutralization-resistant population of viruses. Conclusions: Phylogenetic, evolutionary and selective pressure analysis contributed to improve the knowledge on the circulation of ZIKV.
ARTICLE INFO
Article history:
Received 15 January 2016
Received in revised form 16 February 2016
Accepted 15 March 2016
Available online 20 May 2016
Zika virus spreading in South America: evolutionary analysis of emerging neutralizing resistant Phe279Ser strains
Marta Giovanetti1,2,3*, Teresa Milano4, Luiz Carlos Alcantara3, Laura Carcangiu1, Eleonora Cella1,5, Alessia Lai6, Alessandra Lo Presti1, Stefano Pascarella4, Gianguglielmo Zehender6, Silvia Angeletti7, Massimo Ciccozzi1,81Department of Infectious Parasitic and Immunomediated Diseases, National Institute of Health, Rome, Italy
2Department of Biology, University of Rome ‘Tor Vergata’, Rome, Italy
3Laboratory of Hematology, Genetic and Computational Biology, Gonçalo Moniz Research Center, Oswaldo Cruz Foundation (LHGB/CPqGM/ FIOCRUZ), Salvador, Bahia, Brazil
4Dipartimento di Scienze Biochimiche ‘A. Rossi Fanelli’, Università La Sapienza, 00185 Roma, Italy
5Department of Public Health and Infectious Diseases, Sapienza University of Rome, Rome, Italy
6Laboratory of Infectious Diseases and Tropical Medicine, University of Milan
7Clinical Pathology and Microbiology Laboratory, University Hospital Campus Bio-Medico of Rome, Rome, Italy
8University Campus Bio-Medico, Rome, Italy
ABSTRACT
Objective: To investigate the genetic diversity of Zika virus (ZIKV) and the relationships existing among these circulating viruses worldwide. To evaluate the genetic polymorphisms harboured from ZIKV that can have an infl uence on the virus circulation. Methods: Three diff erent ZIKV dataset were built. The fi rst dataset included 63 E gene sequences, the second one 22 NS3 sequences and the third dataset was composed of 108 NS5 gene sequences. Phylogenetic and selective pressure analysis was performed. The edited nucleic acid alignment from the Envelope dataset was used to generate a conceptual translation to the corresponding peptide sequences through UGene software. Results: The phylogeographic reconstruction was able to discriminate unambiguously that the Brazilian strains are belonged to the Asian lineage. The structural analysis reveals instead the presence of the Ser residue in the Brazilian sequences (however already observed in other previously reported ZIKV infections) that could suggest the presence of a neutralization-resistant population of viruses. Conclusions: Phylogenetic, evolutionary and selective pressure analysis contributed to improve the knowledge on the circulation of ZIKV.
ARTICLE INFO
Article history:
Received 15 January 2016
Received in revised form 16 February 2016
Accepted 15 March 2016
Available online 20 May 2016
Keywords:
1. Introduction
Zika virus (ZIKV) is an emerging mosquito-borne Flavivirus related to dengue, yellow fever, Japanese encephalitis, and West Nile viruses [1] . The genome of ZIKV is a single-stranded RNA of positive polarity of approximately 11 kb. Both ends of the genome contain the 5’ and the 3’untranslated region, which do not encode for viral proteins. The encoded polyprotein is translated and co- and posttranslationally processed by viral and cellular proteases into three structural proteins: capsid (C), premembrane (prM) or membrane (M), and envelope (E); seven non-structural proteins: NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5. The NS5 protein is constituted by two distinct domains, an N-terminal methyltransferase and a C-terminal RNA-dependent RNA polymerase that are required for capping and synthesis of the viral RNA genome, respectively [2,3] .
The virus is primarily transmitted through the bite of infected mosquitoes. It has been isolated from Aedes africanus, Aedes apicoargenteus, Aedes luteocephalus, Aedes aegypti (Ae. aegypti), Aedes vitattus, and Aedes furcifer mosquitoes. Aedes hensilii was the predominant mosquito species found during the Yap islands outbreak in 2007[4,5].
ZIKV has been documented in 1947, when it was isolated, for the fi rst time, from a sentinel rhesus monkey stationed on a tree platform in the Zika forest, Uganda[6]. Since then, epizootics and small epidemics have occurred in Africa and Asia [7] until 2007 whena Zika fever epidemics took place in Yap Island, Micronesia[8].
ZIKV infection requires a differential diagnosis with other infectious such as Dengue and Chikungunya. After a short incubation period of few days, the symptoms appear and usually last from three to 12 d.
The symptoms are arbovirus-like can rash, fever, arthralgia, conjunctivitis, headache, vomiting and edema. The disease is acute but self-limiting. Frequently, the infection course can be asymptomatic[9,10]. The treatment is symptomatic, combining acetaminophen and antihistaminic drugs. Prevention against the infection, since there is no vaccine, relies on individual protection against bites and eradication of mosquitoes (anti-vectorial prevention).
Because of the lack of eff ective vaccines and/or therapies, ZIKV infection, can be considered an emerging disease and consequently a public health issue. In 2013, a large epidemic in French Polynesia was reported concomitantly with a dengue epidemic caused by serotypes 1 and 3. In the early 2015, records of patients presenting a ‘dengue-like syndrome’ appeared in Brazil. A new challenge has arisen in Brazil with the emergence of ZIKV and co-circulation with others arboviruses (i.e., Dengue and Chikungunya virus). Improved surveillance and response measures are needed to mitigate the already burden on health systems in Brazil and limit further spread to other parts of the world.
In the present study, phylogenetic and evolutionary analyses have been performed to investigate the genetic diversity of ZIKV and the relationships existing among these circulating viruses worldwide. As a second aim, we evaluated the genetic polymorphisms harbored from ZIKV that can have an infl uence on the virus circulation.
2. Material and methods
2.1. Datasets
Three diff erent ZIKV dataset were built. The fi rst dataset included 63 E gene sequences, the second one 22 NS3 sequences and the third dataset was composed of 108 NS5 gene sequences. The sequences of all the dataset were downloaded from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). All of these dataset were used to perform the selective pressure analysis. A subset of 57 E gene sequences with known sampling dates and location was built. The sampling dates for the sequences in this subset ranged from 1947 to 2015. The sampling locations were Brazil (BR=6), Cambodia (KH, n=1), Canada (CA=1), Central African Republic (CF=4), Cook Island (CK=1), Côte D’ivoire (CI=11), French Polynesia (PF=1), Gabon (GA=1), Malaysia (MY=1), Micronesia (FM=1), Nigeria (NG=1), Senegal (SN=26) and Uganda (UG=2).
This subset was used, for the presence of the Brazilian strains, to trace the demographic history and the phylogeography reconstruction related to the current epidemic in this South American country never reported before.
2.2. Likelihood mapping
The phylogenetic signal in a data set of aligned DNA or amino acid sequences can be investigated with the likelihood mapping method by analyzing groups of four sequences, randomly chosen, called quartets [11] . For a quartet, just three unrooted tree topologies are possible. The likelihood of each topology is estimated with the maximum likelihood method and the three likelihoods are reported as a dot in an equilateral triangle (the likelihood map).
Three main areas in the map can be distinguished: the three corners representing fully resolved tree topologies, i.e. the presence of treelike phylogenetic signal in the data; the center, which represents star-like phylogeny, and the three areas on the sides indicating network-like phylogeny, i.e. presence of recombination or conf licting phylogenetic signals. Extensive simulation studies have shown that>33% dots falling within the central area indicate substantial star-like signal, i.e. a star-like outburst of multiple phylogenetic lineages [11,12] . Likelihood mapping analyses were performed, on each dataset, with the program TREE-PUZZLE by analyzing 10 000 random quartets.
2.3. Bayesian phylogenetic analysis: demographic history and Phylogeographic recostruction
The sequences of all the dataset were aligned by using Clustal X and manually edited by Bioedit, as already described [13] . The evolutionary model was chosen as the best-fitting nucleotide substitution model in accordance with the results of the hierarchical likelihood ratio test implemented in Modeltest software version 3.7 as already described [14] .
Maximum Clade Credibility tree on the subset (57 envelope gene sequences with known sampling date and location), were inferred using a Markov Chain Monte Carlo (MCMC) Bayesian approach implemented on the program BEAST v1.8, under HKY +Γ+ I model estimating the evolutionary rate using both a strict and an uncorrelated log-normal relaxed clock model. As coalescent priors, three parametric demographic models of population growth (constant size, exponential, expansion) and a Bayesian skyline plot (a non-parametric piecewise-constant model) were compared. The best fi tting models were selected by means of a Bayes factor (BF, using marginal likelihoods) implemented in Beast v 1.8 [16] . In accordance with Kass and Raftery [17] the strength of the evidence against H0 (null hypothesis) was evaluated as follows: 2lnBF<2=no evidence; 2-6=weak evidence; 6-10=strong evidence; and>10=very strong evidence. A negative 2lnBF indicates evidence in favor of H0. Only values of≥6 were considered significant. The MCMC chains were run for at least 50 million generations, and sampled every 5,000 steps. Convergence was assessed on the basis of the eff ective sampling size. Only eff ective sampling size values of>250 were accepted. Uncertainty in the estimates was indicated by 95% highest posterior density (95% HPD) intervals. Statistical support for specif ic clades was obtained by calculating the posterior probability of each monophyletic clade.
The continuous time Markov Chain process over discrete sampling locations implemented in BEAST 1.8[19] was used for the phylogeographical analysis by implementing the Bayesian Stochastic Search Variable Selection model, which allows diff usion rates to be zero with a positive prior probability. The maximum clade credibility tree (the tree with the largest product of posterior clade probabilities) was selected from the posterior tree distribution after a 10% burn-in using the Tree Annotator program version 1.8. The fi nal trees weremanipulated in FigTree version 1.4.2 for display purposes.
The demographic history was analyzed on the subset by performing the Bayesian skyline plot.
2.4. Selective pressure analysis
The CODEML program implemented in the PAML 3.14 software package (http://abacus.gene.ucl.ac.uk/software/ paml.html) was used to investigate the adaptive evolution of the Zika Virus genes (E, NS3 and NS5). The sequences alignments of the three dataset were used to test whether they were under positive selection.
Six models of codon substitution: M0 (one-ratio), M1a (nearly neutral), M2a (positive selection), M3 (discrete), M7 (beta), and M8 (beta and omega) were used in this analysis [20] . Since these models are nested, we used codon-substitution models to f it the model to the data using the likelihood ratio test [21] . The discrete model (M3), with three dn/ds (ω) classes, allows ω to vary among sites by defi ning a set number of discrete site categories, each with its own ω value. Through maximum-likelihood optimization, it is possible to estimate the ω and P values and the fraction of sites in the aligned data set that falls into a given category. Finally, the algorithm calculates the a posteriori probability of each codon belonging to a particular site category. Using the M3 model, sites with a posterior probability exceeding 90% and a p value>1.0 were designated as being ‘positive selection sites’[20]. The site rate variation was evaluated comparing M0 with M3, while positive selection was evaluated comparing M1 with M2. The Bayes empirical Bayes approach implemented in M2a and M8 was used instead to determine the positively selected sites by calculating the posterior probabilities (P) of ω classes for each site [22] . It is worth noting that PAML likelihood ratio tests have been reported to be conservative for short sequences (e.g. positive selection could be underestimated), although the Bayesian prediction of sites under positive selection is largely unaff ected by sequence length[21,23]. The dN/dS rate (ω) was also estimated by the ML approach implemented in the program HyPhy [24] . In particular, the global (assuming a single selective pressure for all branches) and the local (allowing the selective pressure to change along every branch) models were compared by likelihood ratio test. Site-specifi c positive and negative selection were estimated by two dif ferent algorithms: the fi xed-eff ects likelihood, which fi ts an p rate to every site and uses the likelihood ratio to test if dN=dS; and random ef fect likelihood, a variant of the Nielsen-Yang approach [25] , which assumes that a discrete distribution of rates exists across sites and allows both dS and dN to vary independently site by-site. The two methods have been described in more detail elsewhere[26]. In order to select sites under selective pressure and keep our test conservative, a P value of≤0.1 or a posterior probability of≥0.9 as relaxed critical values[26] was assumed.
2.5. Protein sequence and structure analysis
The edited nucleic acid alignment from the Envelope dataset was used to generate a conceptual translation to the corresponding peptide sequences through UGene software[27]. The reference sequence of the ZIKV Envelope protein (Reference Sequence accession: NC_012532.1) for its entire lengths was then aligned, using ClustalX, to the translated protein alignment. Candidate templates for homology modeling, with the reference sequence ZIKV Envelope as a query, were assessed through the Phyre v2.0 server for protein fold recognition[28] and selected on the basis of query coverage, E-value and better quality of the template structures. The model of ZIKV Envelope protein was built based on the known structures of the homologous Envelope proteins from the Japanese Encephalitis virus (PDB ID: 3P54) and from the Dengue virus type 3 (Dt3) (PDB ID: 1UZG). Clustal X calculated the alignment of the target sequence with the selected templates. A total of ten homology models was generated and optimized using Modeller 9.13 [29] . The model with the best values of the Modeller scoring function was chosen for subsequent analysis. To remove unfavorable contacts of amino acid side chains, derived from the homology modeling process, energy minimization was applied to the selected model using the GROMOS96 force-f ield implementation in Swiss-PDB Viewer software (version 4.0.1) [30] . The model was validated with standard programs such as Prosa栻 [Wiederstein et al., 2007] and Procheck [31] . Residue conservation was evaluated through the Consurf server [32] . Alignment display and editing relied on UGene or Jalview [33] programs. Sequence Logos were drawn using WebLogo [34] . Protein structure analysis, in silico mutagenesis and mapping of the single point amino acid substitutions of the Brazilian Envelope protein sequences onto the three dimensional model and fi gure design were performed using PyMOL[35].
3. Results
3.1. Likelihood mapping
The phylogenetic noise of the three different dataset was investigated by means of likelihood mapping. The percentage of dots falling in the central area of the triangles was 6.9% for the fi rst dataset (Envelope gene), 1.7% for the second dataset (NS3 gene) and 15.8% for the third dataset (NS5 gene); as none of the dataset showed more than 33% of noise, all of them contained suff i cient phylogenetic signal.
3.2. Bayesian phylogenetic analysis: demographic history and phylogeografic recostruction
The BF analysis for the subset showed that the relaxed clock fi tted the data signifi cantly better than the strict clock (2 lnBF=15.02 for relaxed clock). Under the relaxed clock the BF analysis showed that the skyline model was better than the other models (2 lnBF>24.568). The estimated mean value of the Zika virus E gene evolutionary rate was of 4.04×10-4substitution/site/year (95% HPD: 1.32×10-4-7.41×10-4).
Figure 1 showed the Bayesian phylogeographic tree of the E gene (subset) that was constructed using the evolutionary rate estimated. Phylogeographic reconstruction showed that the highest state probability for the root of the tree was an African state, as already described (state probability, SP=0.28). Phylogeographic reconstruction showed three statistically supported clade (Clade A, B, C), originated in Senegal (Clade A, SP=0.51) in Malaysia (Clade B, SP=0.30), in Cô te D'ivoire (Clade C, SP=0.46), which inside of them some clusters statistically supported appear.
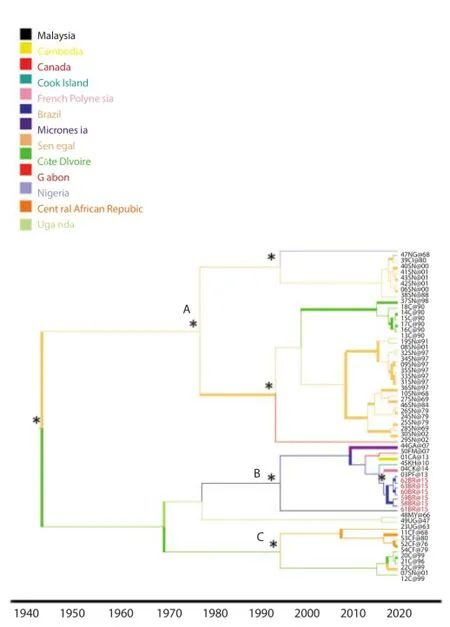
Figure 1. Bayesian phylogeographic tree of Zika virus E gene sequences. The branches are coloured on the basis of the most probable state location of the descendent nodes.
Phylogeographic reconstruction was able to determine that the ZIKV virus outbreak has a single origin and is associated with the Asian phylogenetic clade. Our results are in line with a recent paper, which suggests that the single origin of the ZIKV outbreak in Brazil is from French Polynesia[36].
The demographic history of the fi rst dataset of Zika Virus suggest that the epidemic showed a slight exponential growth up to 2 000 of the epidemic when started a decreasing phase showing a typical ‘bottle neck’ (Figure 2).

Figure 2. Bayesian skyline plot of the fi rst dataset E gene of Zika virus.
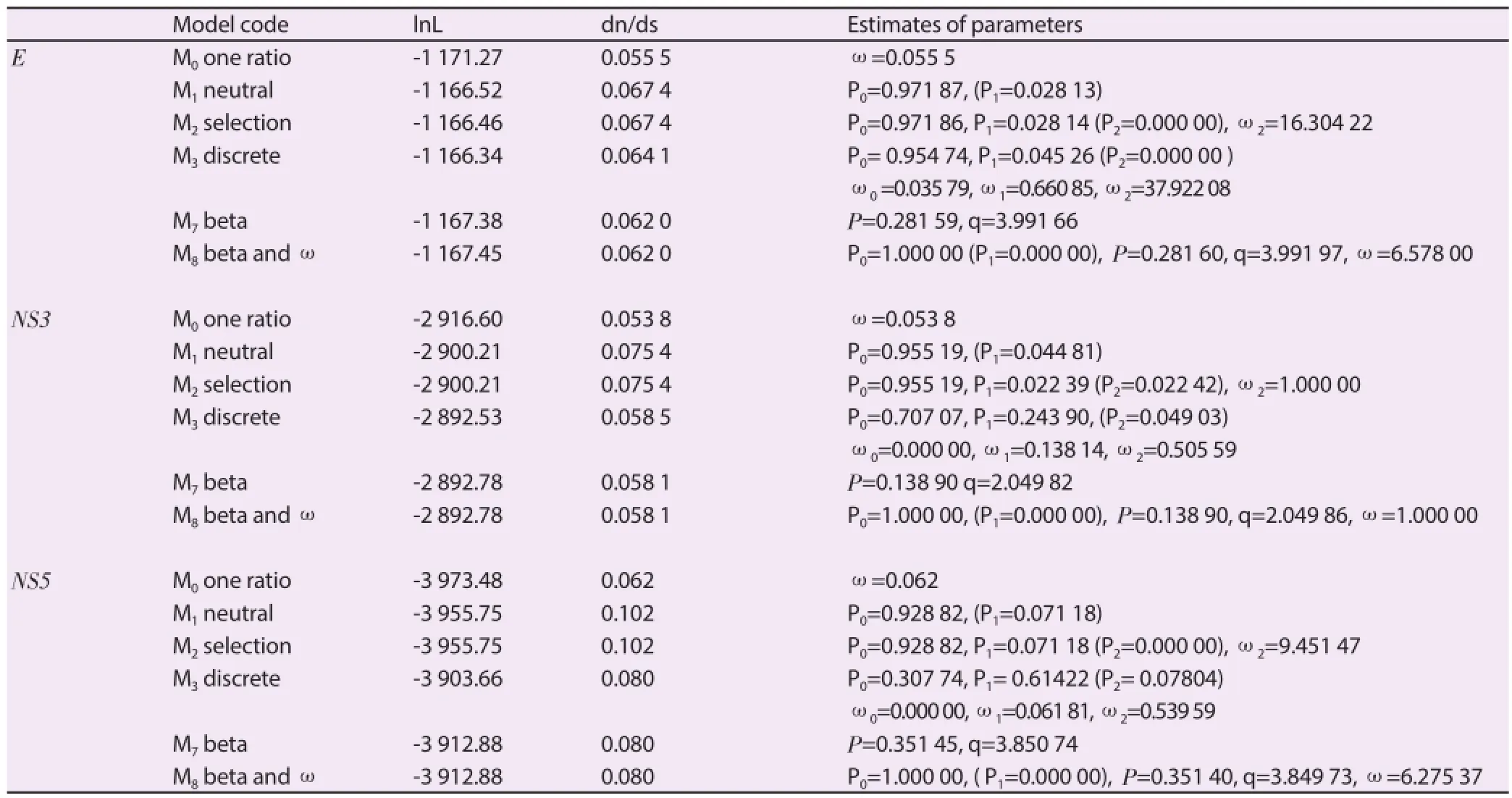
Table 1Likelihood values and parameters estimates for the selection analysis of the E and NS3 and NS3 gene.
3.3. Evolutionary analysis
Selection pressure analyses performed in all the three diff erent datasets E, NS3 and NS5 genes uncovered several sites (59.57%, 49.81% and 39.55% for E, NS3 and NS5 datasets, respectively)under strong negative selection (by using HYPHY) indicated byω(0 suggesting high degree conservation in the genomic region analyzed.
Likewise, the lack of positively selected sites, (both by using HYPHY and PAML), indicated byω(0, is typical of highly adapted phenotypes and shows no detectable directional change on the available data. Likelihood values and parameter estimates obtained from diff erent data sets are listed in Table 1. Estimates of the transition/transversion rate ratio (ts/tv) are quite homogeneous among models in each data set and thus are not shown in Table 1. The average ω ratio ranged from 0.055 5 to 0.067 4 among all models, for E gene dataset, suggesting that a non-synonymous mutation has only 5.55%-6.74
3.4. Protein sequence and structure analysis
The alignment of Envelope proteins pointed out two sites where a residue substitution in the Brazilian sequences with respect to the others can be observed. The Phe in position 279 (numbering refers to the complete ZIKV reference sequence) is replaced by a Ser, while in position 311 an Ile is observed in place of a Val. As in the Brazilian sequence, both substitutions are present in other Envelope protein sequences: more precisely, in the sequences from virus strains isolated in Canada, Cambodia, Malaysia, French Polynesia, Micronesia and Cook Islands (Figure 3A). Except the Malaysian sequence, all the others date back to the last decade.

Figure 3. Mapping of residue characteristic of Brazilian sequences on the Envelope protein of ZIKV A.
Residue conservation analysis carried out through Consurf site, revealed that residues occupying position 279 and 311 of ZIKV reference sequence are not conserved throughout protein sequences of Envelope proteins, independently from their origin (however, it should be noted that only sequences from flavivirus were selected). These amino acidic positions were mapped onto the threedimensional structure of the Envelope protein homology model. superposed to the original template from Dengue type 3 (Figure 3B). The side chains of both residues are located on the surface of the E protein, and probably for this reason they are predicted to be not essential for the maintenance of appropriate protein fold (i.e. the mutations do not destabilize the structure).
Ser 279 is located at the boundary between the so-called Domain Ⅰ(DⅠ), the central domain, and the Domain Ⅱ (DⅡ) (Figure 3B). In the template structures, a Phe residue, as well as in most of ZIKV Envelope proteins, occupies the same position.
Comparison of the amino acid sequences of other flavivirus E proteins, carried out with Consurf analysis, indicated that Phe279 was conserved in most wild-type viruses, although surrounding residues varied. The transition from a hydrophobic to a polar residue, as it is the case of Brazilian sequences, alter the local charge of the pocket: indeed, manual inspection of the structure showed a polar contact (Figure 3C), not observed in the wild type structure, between Ser side chain and the peptide carbonyl oxygen of a conserved Glu residue.
Regarding the other position that appears to be characteristic of Brazilian E sequences, Ile311Val, it should be observed that the position is highly variable in the sequence alignment obtained with other flavivirus sequences. Indeed, looking at the template structure, the structurally equivalent position in Japanese Encephalitis virus and D3t Envelope are occupied by an Asn and a Glu residue, respectively. Moreover, inspection of the model revealed that the Val side chain is projected on the surface of the protein (Figure 3B) in a loop located in the Domain Ⅲ, near to the interface with the other monomer of E protein. So far, no functional role was confi rmed for this residue from previous studies.
4. Discussion
The application of high-resolution phylogenetic methods, such as the Bayesian statistical inference framework, can allow the reconstruction of the geographic history of the still ongoing and never reported before epidemic in Brazil on the basis of the fi rst isolates sampled at known times. Phylogeographic analysis may contribute to better understand the epidemiological history, the diff usion routes of this new epidemic and may contribute to the planning of prevention strategies [37] .
Only few data are actually available regarding the phylogenies of Zika Virus in Brazil. Moreover there are limited sequences available in NCBI especially with regard to the still ongoing epidemic wave in this South American Country. In the present study the phylogenetic analysis was conducted including all sequences with location, available in GenBank.
This is the first study that focusing on Brazilian strains, by the evolutionary and phylogeographic analysis of ZIKV, for the E gene.The phylogeographic analysis showed that, Zika Virus was probably originated in Africa and spread following diff erent routes to the other locations, including African (Senegal, Côte D'ivoire and Uganda) and Asian (Malaysia, Micronesia, French Polynesia) regions. The analysis confi rmed the proposed designation of the two probable lineage Asian and African, that was fi rst reported in 2012 [38] .
For the fi rst time, in this paper, the phylogeographic reconstruction was able to determine that the current ZIKV virus outbreak in Brazil has a single origin and is associated with the Asian phylogenetic clade. Our results seem also to be in line with a recent paper, which suggests that the single origin of the ZIKV outbreak in Brazil is from French Polynesia[36].
The phylodinamic analyses showed a slight exponential growth up to 2 000 of the epidemic when started a decreasing phase showing a typical ‘bottle neck’ that could be explained with measures taken to reclaim[39].
Insecticide treatments have probably driven a selection on the population causing severe bottlenecks and the appearance of insecticide resistance in Ae. aegypti [40,41] .
The genetic polymorphisms of ZIKV may also influence the virus circulation. Virus infectivity and antigenic variability and in particular, antigenic variation may play an important role in the ability of these viruses to escape the host immune response. In our study, using a selective pressure analysis method, negatively selected sites, were mostly found, suggesting the stability of the viral E, NS3 and NS5 proteins.
The alignment of Envelope proteins pointed out two sites where a residue substitution in the Brazilian sequences with respect to the others can be observed.
Ser 279 is located at the boundary between the so called Domain 栺, the central domain, and the Domain栻, that contribute to important dimerization contacts that coordinate the antiparallel E arrangement on mature virus particle [42] . This region, defi ned as the ‘kl’ beta-hairpin binding pocket, has a role in the conformational rearrangement that drives membrane fusion [43] . In the template structures, a Phe residue, as well as in most of ZIKV Envelope proteins, occupies the same position. Comparison of the amino acid sequences of other flavivirus E proteins, carried out with Consurf analysis, indicated that Phe279 was conserved in most wild-type viruses, although surrounding residues varied. This conservation is in line with the function of the hydrophobic pocket, which through a conformational shift, is able to accept hydrophobic ligands [44] . The transition from a hydrophobic to a polar residue, as it is the case of Brazilian sequences, alter the local charge of the pocket: indeed, manual inspection of the structure showed a polar contact, not observed in the wild type structure, between Ser side chain and the peptide carbonyl oxygen of a conserved Glu residue. A similar Phe to Ser substitution in the equivalent position of the E protein of Dengue 3 virus was reported by Lee et al.[45]: they observed that this mutation causes escape from neutralization with IgM M10 and was associated with altered pH sensitivity of that virus. Moreover, the substitution of Ser for Phe at E279 of the dengue 1 neutralizationresistant virus population was demonstrated to be a nonconservative change that increased the hydrophilicity of this region of the protein [46] . A sort of analogy can be do with chikungunya virus about the A226V of the E1 protein. Lo Presti et al.[47] followed this variant reconstructing the geographic spread of CHIKV during the last epidemic wave. This mutation was important and necessary to change the chikungunya vector from Ae. aegypti to Aedes Albopictus determining the Indian Ocean outbreak [47] . Because only few sequences have been available from Brazil it is important to follow and to confirm the eventual introduction of ZIKV in Brazil from Asian regions. A more extensive analysis of additional samples from other Brazilian regions, as well as a complete viral genetic characterization is needed.
All these observations suggest that, also in ZIKV Envelope, the region around Phe279Ser mutation might act as a hinge for low pH-induced conformational changes accompanying the E protein dimer to trimer transition, which occurs prior to its fusion with host cell membranes. The presence of the Ser residue in the Brazilian sequences (however already observed in other previously reported ZIKV infections) could suggest the presence of a neutralizationresistant population of viruses [48] .
In conclusion, the study trough the phylogeographic and selective pressure analysis contributed to improve the knowledge on the circulation of ZIKV in Brazil. From these analysis emerged also the indication of a possible outbreak in Brazil with strains Phe279Ser neutralizing resistant that could indicate the need of a molecular epidemiological monitoring.
The understanding of the epidemiology of ZIKV is limited and the evolution of the outbreak needs to be carefully investigated to better assess the risk of spread and its consequences for public health. The knowledge of the circulating ZIKV lineages in Brazil is considered essential, as the Asian lineage seems to have a high epidemic potential.
Conflict of interest statement
We declare that we have no confl ict of interest.
References
[1] World Health Organization (WHO). Zika virus outbreaks in the Americas. Wkly Epidemiol Rec 2015; 90: 609-610.
[2] Chambers TJ, Chang HS, Galler R, Rice CM. Flavivirus genome organization, expression and replication. Annu Rev Microbiol 1990; 44: 649-688.
[3] Kuno G, Chang GJJ. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 2007; 152: 687-696.
[4] Hayes EB. Zika virus outside Africa. Emerg Infect Dis 2009; 15: 1347-1350.
[5] Marcondes CB, Ximenes MF. Zika virus in Brazil and the danger of infestation by Aedes (Stegomyia) mosquitoes. Rev Soc Bras Med Trop 2015; Dec 22. http://dx.doi.org/10.1590/0037-8682-0220-2015.
[6] Dick GW, Kitchen SF, Haddow AJ. Zika virus栺. Isolations and serological specifi city. Trans R Soc Trop Med Hyg 1952; 46: 509-520.
[7] Hayes EB. Zika virus oitside Africa. Emerg Infect Dis 2009; 15: 1347-1350.
[8] Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 2009; 360: 2536-2543.
[9] Grard G. Zika virus in Gabon (Central Africa)-2007: a new threat from Aedes albopictus? PLoS Negl Trop Dis 2014; 8: e2681.
[10] Gatherer D, Kohl A. Zika virus: a previously slow pandemic spreads rapidly through the Americas. J Gen Virol 2016; 97(2): 269-273.
[11] Strimmer K, von Haeseler A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc Natl Acad Sci 1997; 94: 6815-6819.
[12] Salemi M, de Oliveira T, Ciccozzi M, Rezza G, Goodenow MM. Highresolution molecular epidemiology and evolutionary history of HIV-1 subtypes in Albania. PLoS One 2008; 3(1): e1390.
[13] Ciccozzi M, Vujošević D, Lo Presti A, Mugoša B, Vratnica Z, Lai A, et al. Genetic diversity of HIV type 1 in Montenegro. AIDS Res Hum Retroviruses 2011; 27: 921-924.
[14] Ciccozzi M, Lai A, Ebranati E, Gabanelli E, Galli M, Mugosa B, Vratnica Z, et al. Phylogeographic reconstruction of HIV type 1B in Montenegro and the Balkan region. AIDS Res Hum Retroviruses 2012; 28: 1280-1284.
[15] Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, Diallo M, et al. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis 2014; 8(1): e2636.
[16] Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 2005; 22: 1185-1192.
[17] Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc 1995; 90: 773-795.
[18] Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007; 7: 214.
[19] Lemey P, Rambaut A, Drummond AJ, Suchard MA. Bayesian phylogeography fi nds its roots. PLoS Comput Biol 2009; 5: 1-16.
[20] Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000; 155: 431-449.
[21] Anisimova M, Bielawsky JP, Yang Z. Accuracy and power of likelihood ratio test in detecting adaptive evolution. Mol Biol Evol 2001; 18: 1585-1592.
[22] Yang Z, Wong WS, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 2005; 22: 1107-1118.
[23] Anisimova M, Bielawsky JP, Yang Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol 2002; 19: 950-958.
[24] Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005; 21: 676-679.
[25] Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998; 148(3): 929-936.
[26] Kosakovsky Pond SL, Frost SD. Not so diff erent after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 2005; 22: 1208-1222.
[27] Okonechnikov K, Golosova O, Fursov M. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 2012; 28(8): 1166-1167.
[28] Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modelling, prediction and analysis. Nat Protoc 2015; 10: 845-858.
[29] Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993; 234: 779-815.
[30] Kaplan W, Littlejohn TG. Swiss-PDB viewer (deep view). Brief Bioinform 2001; 2: 195-197.
[31] Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 1996; 8: 477-486.
[32] Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 2010; 38 (suppl 2): W529-W533.
[33] Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009; 25: 1189-1191.
[34] Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome research 2004; 14: 1188-1190.
[35] Schroedinger L. The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC. 2015.
[36] Musso D. Zika Virus Transmission from French Polynesia to Brazil. Emerg Infect Dis 2015; 21: 1887.
[37] Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 2009; 10: 540-550.
[38] Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, et al. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis 201; 6: e1477.
[39] Herrera F, Urdaneta L, Rivero J, Zoghbi N, Ruiz J, Carrasquel G, et al. Population genetic structure of the dengue mosquito Aedes aegypti in Venezuela. Mem Inst Oswaldo Cruz 2006; 101: 625-633.
[40] Bisset JA, Rodriguez MM, Molina D, Diaz C, Soca LA. High esterases as mechanism of resistance to organophos-phate insecticides in Aedes aegypti strains. Rev Cubana Med Trop 2001; 53: 37-43.
[41] Rodrí guez MM, Bisset J, de Ferná ndez DM, Lauzan L, Soca A. Detection of insecticide resistance in Aedes aegypti (Diptera: Culicidae) from Cuba and Venezuela. J Med Entomol 2001; 38: 623-628.
[42] Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995; 375: 291-298.
[43] Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004; 427: 313-319
[44] Modis Y, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci 2003; 100: 6986-6991.
[45] Lee E, Weir RC, Dalgarno L. Changes in the dengue virus major envelope protein on passaging and their localization on the threedimensional structure of the protein. Virology 1997; 232: 281-290.
[46] Beasley DW, Aaskov JG. Epitopes on the dengue 1 virus envelope protein recognized by neutralizing IgM monoclonal antibodies. Virology 2001; 279: 447-458.
[47] Lo Presti A, Ciccozzi M, Cella E, Lai A, Simonetti FR, Galli M, et al. Origin, evolution, and phylogeography of recent epidemic CHIKV strains. Infect Genet Evol 2012; 12: 392-398.
[48] Pierson TC, Kielian M. Flaviviruses: braking the entering. Curr Opin Virol 2013; 3: 3-12.
E-mail: ciccozzi@iss.it
Tel: +390649903187
Zika virus
Phylogeny
Evolution
doi:Document heading 10.1016/j.apjtm.2016.03.028
*Corresponding author:Massimo Ciccozzi, Department of Infectious Parasitic and Immunomediated Diseases, Reference Centre on Phylogeny, Molecular Epidemiology and Microbial Evolution (FEMEM)/ Epidemiology Unit, National Institute of Health, Rome, Italy.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Study on the role of Cathepsin B and JNK signaling pathway in the development of cerebral aneurysm
- Influence of hydrogen sulfide on zymogen activation of homocysteineinduced matix metalloproteinase-2 in H9C2 cardiocytes
- Effect and mechanism of miR-34a on proliferation, apoptosis and invasion of laryngeal carcinoma cells
- Mechanism of TLR-4/NF-κB pathway in myocardial ischemia reperfusion injury of mouse
- Exploration of the regulatory effect of miR-21 on breast cancer cell line proliferation and invasion as well as the downstream target genes
- Serological survey on some pathogens in wild brown hares (Lepus europaeus) in Central Italy