Spin Polarization at Organic-Ferromagnetic Interface:Effect of Contact Configuration
2016-07-05YingLiGuangpingZhangZhenXieZhaoZhangJunfengRenChuankuiWangGuichaoHuSchoolofPhysicsandElectronicsShandongNormalUniversityJinan250014China
Ying Li,Guang-ping Zhang,Zhen Xie,Zhao Zhang,Jun-feng Ren,Chuan-kui Wang,Gui-chao HuSchool of Physics and Electronics,Shandong Normal University,Jinan 250014,China
Spin Polarization at Organic-Ferromagnetic Interface:Effect of Contact Configuration
Ying Li,Guang-ping Zhang,Zhen Xie,Zhao Zhang,Jun-feng Ren,Chuan-kui Wang,Gui-chao Hu∗
School of Physics and Electronics,Shandong Normal University,Jinan 250014,China
(Dated:Received on November 11,2015;Accepted on January 25,2016)
Based on ab initio theory,the interfacial spin polarization of a benzene-dithiolate molecule vertically adsorbed on a nickel surface is investigated by adopting different microscopic contact configurations. The results demonstrate a strong dependence of the interfacial spin polarization on the contact configuration,where the sign of spin polarization may vary from positive to negative with the change of contact configuration. By analyzing the projected density of states,an interfacial orbital hybridization between the 3d orbital of the nickel atom and the sp3hybridized orbital of the sulfur atom is observed. We also simulated the interfacial adsorption in mechanically controllable break junction experiments. The magnetoresistance obtained from Julliere model is about 27%based on the calculated interfacial spin polarization,which is consistent with experimental measurement.
Key words:Organic spintronics,Spin polarization,Interface,Contact configuration
∗Author to whom correspondence should be addressed. E-mail:hgc@sdnu.edu.cn
I. INTRODUCTION
Spintronics has developed rapidly in past decades which designs functional devices with the utilization of electron spin[1]. Recently,one of the stirring progresses in this field is the rise of molecular spintronics,which combines both the advantages of spintronics and organic materials[2-4]. Spin-orbital interaction in organic molecules is very weak since most organic molecules are composed of light elements. This induces a long spin relaxation time in organic materials which is advantageous for spin memory and transport. Many organic spintronic devices have been widely designed and tested to now,such as organic spin valves[5,6]and organic spin filters[7,8].
In organic spintronic devices,the interfaces between organic molecules and ferromagnetic electrodes are particularly important in determining the performance of the device. For example,Xiong et al. constructed La0.7Sr0.3MnO3(LSMO)/Alq3/Co structure of organic spin valves and discovered a negative magnetoresistance (MR)about 40%in low temperature[9]. However,the subsequent experimental measurements on the same structure with different techniques demonstrated various MR properties. Barraud et al. used atomic force microscopy(AFM)and measured a positive MR up to 300%[10]. With a buffer layer assisted growth method,Sun et al. also obtained a positive MR[11]. A possible reason for the large deviation of measurements can be tracked from the interfaces formed with different synthesis techniques,such as different shapes of surface or different contact configurations. The understanding of the spin polarization at different interfaces tends to be a key point for spin injection and spin transport in organic spintronic devices.
Unlike inorganic interface,when an organic molecule is adsorbed on a ferromagnetic metal surface,orbital hybridization between the molecule and the ferromagnet may happen,which modifies the interfacial spin polarization distinct from the bulk ferromagnet. Atodiresei et al. found that the interfacial spin polarization may be inversed from the bulk ferromagnet by horizontally adsorbing an aromatic molecule on the Fe surface due to a pz-d exchange mechanism[12]. Mandal and Pati also found that a small change in the interfacial distance at the metal-molecule junction may lead to an inversion of the tunneling magnetoresistance(TMR)[13]. Sanvito and Yi et al. showed that in molecular magnets based devices the spin polarization and spin transport may be tuned by interfacial manipulation[14,15]. Particularly,as the most typical sample,the benzene-based magnetic interfaces and devices are widely investigated with different ferromagnetic metals both in theories and experiments[16-20].
In spite of the above studies,the organic/ferromagnetic interface still needs theoretical attention due to the following reasons:firstly,in most studies a flat adsorption of an aromatic molecule on the ferromagnetic surface was usually considered to stress the pzd coupling between the molecule and the ferromagnet,which is different from the single-molecule spin-valve experiment where the molecule should be possibly coupled to the ferromagnetic surface vertically via sulfur atoms[20]. Secondly,although the single-molecule spin-valve structure has been calculated by several theoretical studies,the microscopic details of the interface were not expanded. In this work,we theoretically studied the interfacial spin polarization by adsorbing a benzenedithiolate(BDT)molecule perpendicular to a nickel (Ni)surface. The effect of microscopic contact configuration,typically including hollow,bridge and top,on the spin polarization was investigated. A special interface was also constructed to simulate the adsorption in MCB junctions.
II. COMPUTATIONAL METHODS
Here a BDT molecule is used in our calculation,where two para-site hydrogen atoms in a benzene ring are replaced by two thiol groups. The molecule is adsorbed on a Ni(111)surface vertically(along z direction)by one sulfur atom with its hydrogen atom detached as shown in Fig.1. A 3×3×3 unit cell is used to simulate the Ni(111)surface(in xy plane)and periodic boundary conditions are used in the x and y directions. The lattice constant for the z direction is set to be large enough so that a Ni(111)slab is present. To investigate the effect of contact configuration,three typical different adsorption structures are considered:the sulfur atom is adsorbed at the top site,the bridge site,and the hollow site respectively,which are shown in Fig.1(b),(c),and (d).
The numerical calculations are performed by using the density-functional theory implemented in SIESTA package[21]. The Perdew-Burke-Ernzerhof(PBE)[22]exchange-correlation functional for generalized gradient approximation(GGA)is adopted. The core electrons are represented by pseudopotentials. A flexible numerical atomic-like wave-functions basis set is used to handle valence electrons[23]. Double-ξ plus single polarization orbitals(DZP)are used for the BDT molecule while single-ξ plus single polarization orbitals(SZP)are used for Ni atoms. These functional and bases have been used to optimize the structure of molecular magnetic contacts successfully before[15]. The finite real-space grid for numerical calculations is defined using a 300 Ry energy cutoff. The geometry of the system is optimized with conjugate gradient method until the atomic forces are less than 0.02 eV/Å. During the relaxation,the anchoring sulfur atom is fixed to the bridge,top or hollow site respectively. After the structural optimization,the spin-dependent density of states(DOS)will be calculated and analyzed.

III. RESULTS AND DISCUSSION
Before studying the interfacial spin polarization,we first calculated the density of state(DOS)of the isolated Ni electrode and the BDT molecule without interaction. Figure 2(a)shows the spin-dependent DOS of the total Ni electrode. Here the majority and the minority correspond to spin-up and spin-down electrons respectively. If we define a spin polarization of the DOS as:

it is found that the spin polarization at the Fermi energy is negative although the spin-up electrons in total is more than the spin-down ones. This is consistent with other first-principle calculation[24]. The DOS of the BDT molecule shown in Fig.2(b)is spin degenerate with an energy gap of 2.8 eV between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO).
In the presence of interaction,a key question is that how the organic molecule is spin polarized in the presence of orbital hybridization between the molecule and the ferromagnetic electrode. In Fig.2(c)-(e),we plot the projected density of states(PDOS)onto the BDT molecule at three different contact configurations. It is found that an obvious spin nondegeneracy of the PDOS is observed around the Fermi level in the[-1.0,1.0]eV energy interval,while the spin splitting of the PDOS away from the Fermi level is not obvious. This is different from flat adsorption of a benzene molecule on the ferromagnetic surface[12],where an apparent spin polarization can be observed in a wide energy range of energy interval.
The spin polarization of the PDOS of the BDT molecule at different contact configurations is calculated. As shown in Fig.3,the plot demonstrates an energy dependent spin polarization at all three cases,which is similar to that of flat adsorption[12]. The spin polarization oscillates between positive and negative values,where a larger amplitude exists in the range of[-4.0,1.0]eV energy interval. The maximum magnitude of the spin polarization appears at around -0.9 eV for all the three configurations,which is about 0.46 for top,0.44 for bridge,and 0.56 for hollow adsorption. Particularly,we notice that the spin polarization in several energy intervals differs from negative to posi-tive with the change of contact configuration,especially around the Fermi level. For example,from -0.6 eV to 0.0 eV,the hollow adsorption corresponds to a positive spin polarization up to 0.4,while for the top position it is negative down to -0.3. This means the interfacial spin polarization strongly depends on the microscopic contact configuration. It should be mentioned that although the hollow adsorption is the most stable among the three configurations by calculating the total energy,the bridge and top adsorptions are still possible due to the interfacial irregularity in experiments,which may induce different spin polarizations as well as spin transport behaviors.
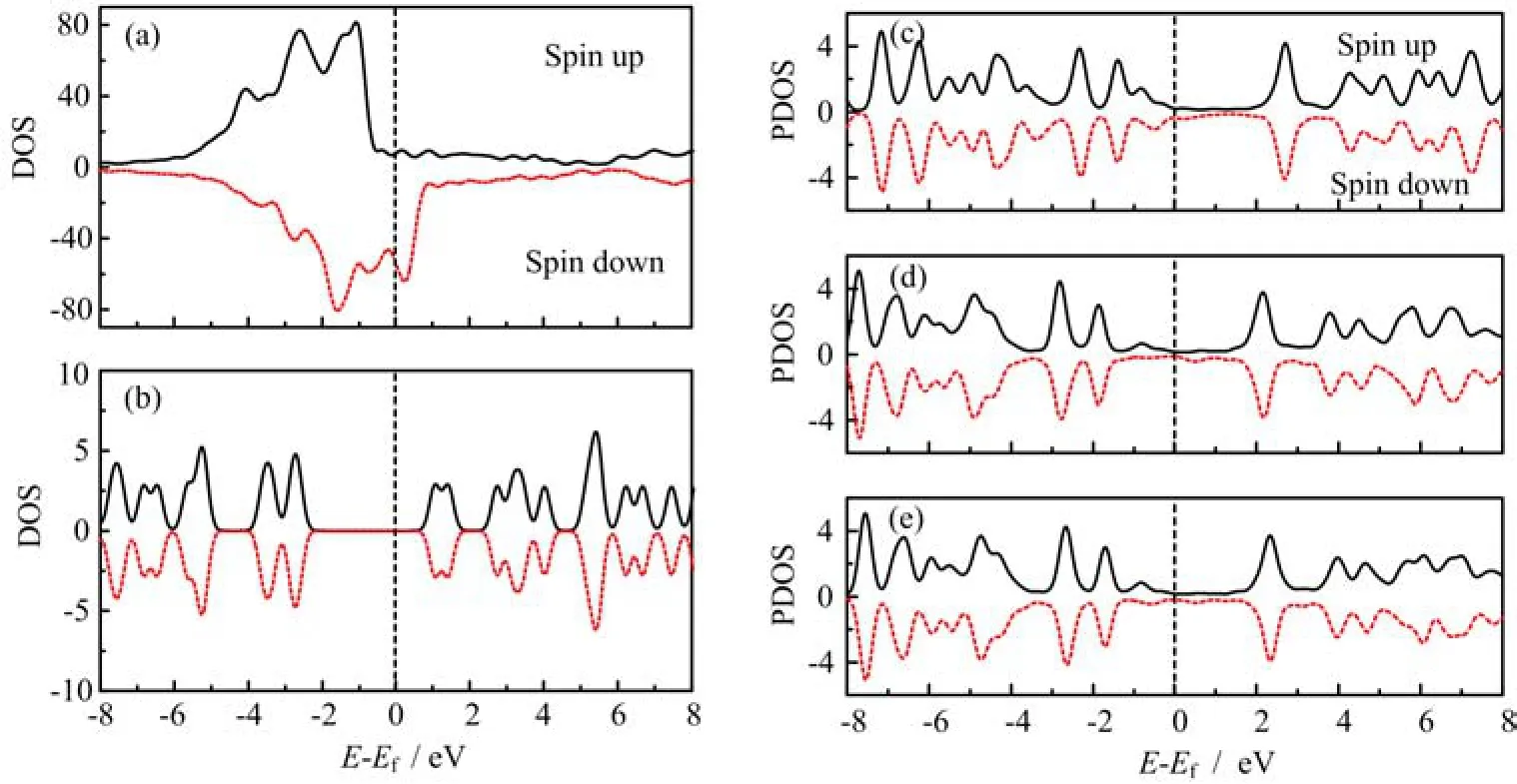
FIG. 2 Density of states of isolated(a)Ni electrode and(b)BDT molecule. The projected density of states on the BDT molecule at three contact configurations including(c)top,(d)bridge,and(e)hollow,with interaction between the Ni electrode and the BDT molecule.
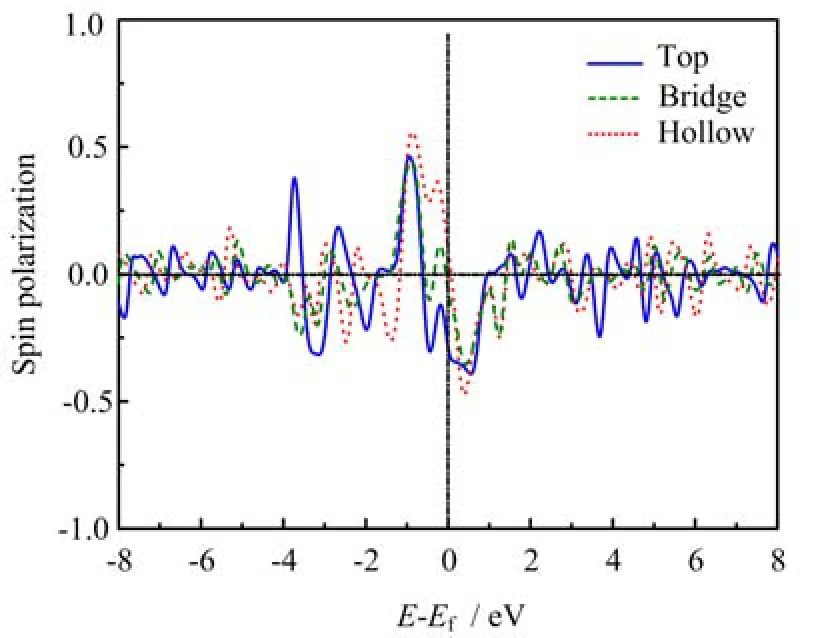
FIG. 3 Spin polarization of the PDOS of the BDT molecule at three different contact configurations.
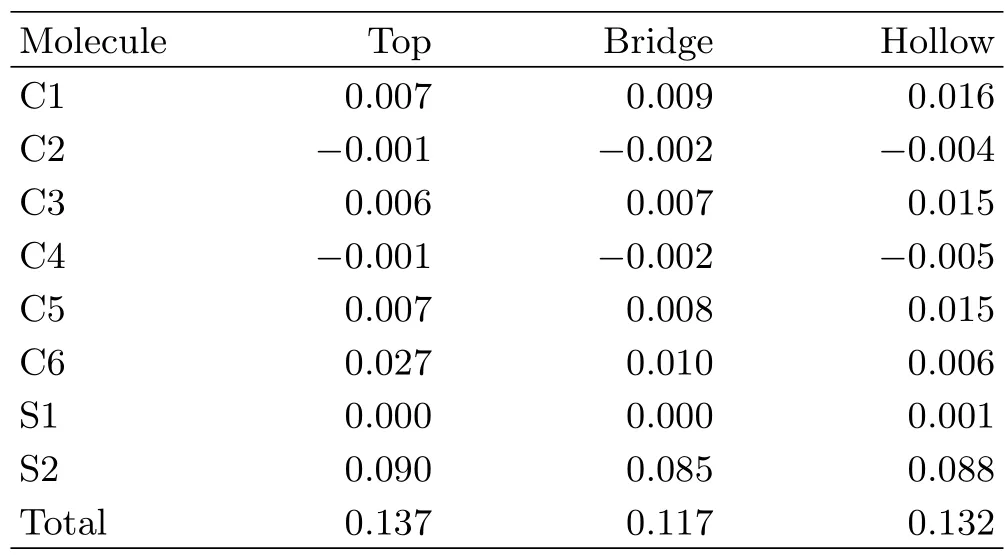
TABLE I Variation of net spin(in unit of ħ/2)on carbon,sulfur and the total molecule after adsorption. Labels of atoms refer to Fig.1(a).
To explore the binding details we first calculate the variation of net spin in the BDT molecule after adsorption. The net spin on each atom is defined as the difference between the numbers of spin-up and spin-down electrons on the atom,which is calculated based on a Bader analysis[25]of the optimized system. Of course the net spin density along the isolated molecule is zero. The results after adsorption at three contact configurations are shown in Table I. It is found that a net spin appears in the BDT molecule after adsorption in all three cases,which are 0.137,0.117 and 0.132 for top,bridge and hollow adsorption,respectively. We also notice that the net spin of the molecule mainly distributes on the sulfur atom close to the Ni surface,which indicates the hybridization mainly happens between the sulfur atom and the coupled Ni atom. This is also verified by checking the change of PDOS onto the adsorbing S atom and the left component in the molecule respectively. We also checked the bond length near the interface at each contact configuration. It is found that the bond length of S-Ni is 3.93,3.77 and 3.76Å respectively for top,bridge and hollow adsorption. This looks reasonable that the longest S-Ni bond appears in the top adsorption,where the orbital hybridization between the molecule and the Ni atoms isthe weakest among the three cases and the interfacial spin polarization at Fermi energy still keeps negative close to that of the electrode. The C-S bond changes little,which is 1.77,1.78 and 1.79Å for top,bridge and hollow adsorption.
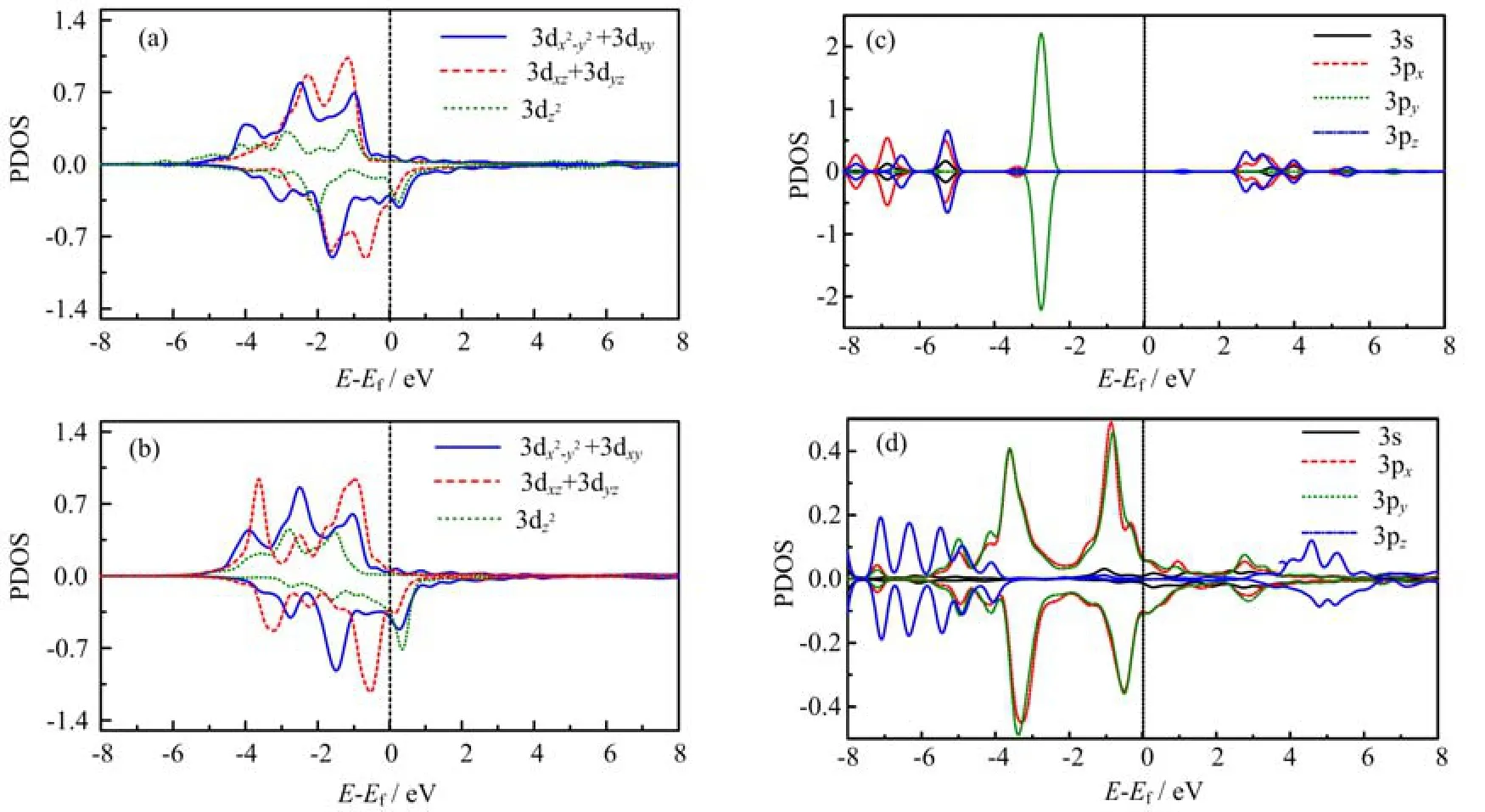
FIG. 4 PDOS of the interfacial interacting Ni and S atoms at the top adsorption.(a)Ni before adsorption,(b)Ni after adsorption,(c)S before adsorption,(d)S after adsorption.
A deep orbital analysis is then performed by investigating the atomic orbital PDOS of the interacting atoms at the interface. Here we take the top adsorption as the sample. Figure 4 compares the atomic orbital PDOS of the adsorbed Ni atom and S atom before and after adsorption. From Fig.4(a)and(b)one can find that the out-of-plane z component of d orbital of the Ni atom is modified and contributes to the orbital hybridization. Furthermore,the hybridization around the Fermi level is contributed by the spin-down states of the Ni. For the S atom,it is found that all the components of 3s and 3p states are disturbed in the hybridization as shown in Fig.4(c)and(d). The spin polarization near the Fermi level mainly comes from the spin nondegeneracy of pxand pyorbitals. This is distinct from the flat adsorption of benzene molecule where the orbital hybridization is contributed by the pzcomponent of p orbital. This is because the terminal S atom in the BDT molecule has a sp3orbital hybridization as that in the SH group. Thus the interacting states with the d orbital of the Ni atom include all the components of the s and p states of the S atom. A similar mechanism of the orbital hybridization is observed in the bridge and hollow adsorption(not shown here). However,the contact angle between the bonds of the Ni and the S atoms as well as the interacting strength will differ with the change of microscopic contact configurations,which may lead to a different interfacial spin polarization.
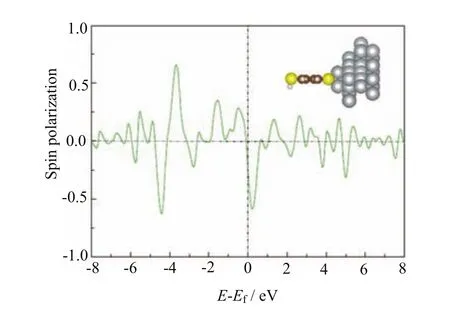
FIG. 5 Spin polarization of the PDOS of the BDT molecule at the configuration of three extra Ni atoms on the surface.
At last,we further construct a special contact configuration which has been adopted to simulate the adsorption in MCB junctions[26,27]. Here three extra Ni atoms are added on the Ni electrode surface to form an equilateral triangle structure. The sulfur atom is adsorbed on the hollow position of the equilateral triangle,as shown in the insert of Fig.5. The BDT molecule including the adsorbed S atom was fully relaxed during the optimization. Here the optimized S-Ni bond is 3.16Å. The calculated energy dependent spin polarization is shown in Fig.5. A similar curve to the top adsorption is observed,where a negative spin polarization is obtained at the Fermi energy. However,the negative spin polarization near the Fermi level achieves almost 60%,which is much larger than that in top adsorption. The spin polarization around -4.0 eV is also enhanced. To achieve a simple comparison with the MCB junctionexperiment,we use the Julliere model[28]to estimate the MR of Ni/BDT/Ni junction based on the interfacial spin polarization in MCB adsorption,
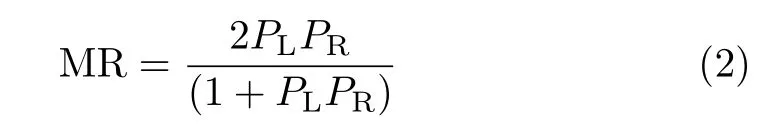
where PLand PRare the left and right interfacial spin polarization. Here for simplicity we assume that the two interfaces are identical as we presented here. In the Ni/BDT/Ni junctions experiment,a 30%MR is measured at the bias voltage of 10 mV[20]. So we use the spin polarization around 10 meV,which is about 40%,to perform calculation. The calculated MR is 28%,which is close to the experimental value. We also noticed that our estimation of MR is similar to other calculation of spin transport through Ni/BDT/Ni with nonequilibrium Greens function method,where a value of 27%for MR is obtained[29].
IV. CONCLUSION
We have performed a theoretical study on the interfacial spin polarization of a BDT molecule vertically adsorbed on a Ni surface. The effect of interfacial contact configuration is focused. By calculating the spin polarization in three different contact configurations,namely top,bridge and hollow,we find that the interfacial spin polarization strongly depends on the contact configuration. The spin polarization is energy dependent,the sign and magnitude vary with the change of contact configuration. The binding mechanism is analyzed from the projected density of states of the molecule and the terminal interacting atoms. The orbital hybridization happens between the 3d orbital of the Ni atom and the sp3hybridized orbital of the terminal S atom. We further simulate the adsorption in MCB junction by adding three extra Ni atoms on the surface. A large negative spin polarization up to 60%is obtained around Fermi level. With the help of Julliere model we estimated the MR of the MCB adsorption based on our interfacial spin polarization. A value of 28%for the MR is obtained,which is consistent with the experimental measurement and other ab intio calculation. This work indicates that the microscopic contact configuration in organic spintronic devices plays an important role in determining the interfacial spin polarization,which can not be neglected during the understanding of the spin transport property.
V. ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.11374195 and No.11405098),the Natural Science Foundation of Shandong Province(No.ZR2014AM017),the Taishan Scholar Project of Shandong Province,and the Excellent Young Scholars Research Fund of Shandong Normal University.
[1]A. Fert,Rev. Mod. Phys. 80,1517(2008).
[2]W. J. M. Naber,S. Faez,and W. G. van der Wiel,J. Phys. D 40,R205(2007).
[3]V. A. Dediu,L. E. Hueso,I. Bergenti,and C. Taliani,Nat. Mater. 8,707(2009).
[4]T. Sugawara and M. M. Matsushita,J. Mater. Chem. 19,1738(2009).
[5]F. Wang and Z. V. Vardeny,J. Mater. Chem. 19,1685 (2009).
[6]G. C. Hu,M. Y. Zuo,Y. Li,J. F. Ren,and S. J. Xie,Appl. Phys. Lett. 104,033302(2014).
[7]J. S. Bhat,R. A. Nesargi,and B. G. Mulimani,Phys. Rev. B 73,235325(2006).
[8]G. C. Hu,Y. Guo,J. H. Wei,and S. J. Xie,Phys. Rev. B 75,165321(2007).
[9]Z. H. Xiong,D. Wu,Z. V. Vardeny,and J. Shi,Nature 427,821(2004).
[10]C. Barraud,P. Seneor,R. Mattana,S. Fusil,K. Bouzehouane,C. Deranlot,P. Graziosi,L. Hueso,I. Bergenti,V. Dediu,F. Petroff,and A. Fert,Nat. Phys. 6,615 (2010).
[11]D. Sun,L. Yin,C. Sun,H. Guo,Z. Gai,X. G. Zhang,T. Z. Ward,Z. Cheng,and J. Shen,Phys. Rev. Lett. 104,236602(2010).
[12]N. Atodiresei,J. Brede,P. Lazic,V. Caciuc,G. Hoffmann,R. Wiesendanger,and S. Blugel,Phys. Rev. Lett. 105,066601(2010).
[13]S. Mandal and R. Pati,ACS Nano 6,3580(2012).
[14]S. Sanvito,Chem. Soc. Rev. 40,3336(2011).
[15]Z. L. Yi,X. Shen,L. L. Sun,Z. Y. Shen,S. M. Hou,and S. Sanvito,ACS Nano 4,2274(2010).
[16]X. Sun,Y. Yamauchi,M. Kurahashi,T. Suzuki,Z. P. Wang,and S. Entani,Chem. Phys. Lett. 452,156 (2008).
[17]Z. Yang,B. L. Zhang,X. G. Liu,Y. Z. Yang,X. Y. Li,S. J. Xiong,and B. S. Xu,Euro. Phys. Lett. 104,50006 (2013).
[18]H. Y. He,R. Pandey,R. Pati,and S. P. Karna,Phys. Rev. B 73,195311(2006).
[19]Q. Zhang,W. B. Mi,X. C. Wang,and X. H. Wang,Sci. Rep. 5,10602(2015).
[20]R. Yamada,M. Noguchi,and H. Tada,Appl. Phys. Lett. 98,053110(2011).
[21]J. M. Soler,E. Artacho,J. D. Gale,A. Garca,J. Junquera,P. Ordejn,and D. Snchez-Portal,J. Phys.:Condens. Matter 14,2745(2002).
[22]M. Ropo,K. Kokko,and L. Vitos,Phys. Rev. B 77,195445(2008).
[23]R. C. Longo,M. M. G. Alemany,J. Ferrer,A. Vega,and L. J. Gallego,J. Chem. Phys. 128,114315(2008).
[24]F. Cinquini,F. Delbecq,and P. Sautet,Phys. Chem. Chem. Phys. 11,11546(2009).
[25]W. Tang,E. Sanville,and G. Henkelman,J. Phys.:Codens. Mater 21,084204(2009).
[26]G. P. Zhang,G. C. Hu,Z. L. Li,and C. K. Wang,J. Phys. Chem. C 116,3773(2012).
[27]G. P. Zhang,G. C. Hu,Y. Song,Z. Xie,and C. K. Wang,J. Chem. Phys. 139,094702(2013).
[28]M. Julliere,Phys. Lett. 54,225(1975).
[29]D. Waldron,P. Haney,B. Larade,A. H. Macdonald,and H. Guo,Phys. Rev. Lett. 96,166804(2006).
DOI:10.1063/1674-0068/29/cjcp1511230
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Impact of Eu3+Ions on Physical and Optical Properties of Li2O-Na2O-B2O3Glass
- Investigation of Ultrafast Electronic Transfer Process on Organic/Inorganic Heterojunction by Femtosecond Transient Absorption
- Ordered Toroid Structures of Nanoparticles in Self-attractive Semiflexible Polymer/Nanoparticle Composites
- Virtual Screening of Human O-GlcNAc Transferase Inhibitors
- Morphology and Growth Process of Bat-like ZnO Crystals by Thermal Evaporation
- Epitaxial Growth and Thermoelectric Measurement of Bi2Te3/Sb Superlattice Nanowires