辛基酚聚氧乙烯醚磺酸盐界面行为的分子动力学模拟
2016-07-04单晨旭曹绪龙祝仰文刘坤曲广淼吕鹏飞薛春龙丁伟东北石油大学化学化工学院黑龙江大庆6338中国石化胜利油田分公司地质科学研究院山东东营5705
单晨旭,曹绪龙,祝仰文,刘坤,曲广淼,吕鹏飞,薛春龙,丁伟(东北石油大学化学化工学院,黑龙江 大庆 6338;中国石化胜利油田分公司地质科学研究院,山东 东营 5705)
辛基酚聚氧乙烯醚磺酸盐界面行为的分子动力学模拟
单晨旭1,曹绪龙2,祝仰文2,刘坤1,曲广淼1,吕鹏飞1,薛春龙1,丁伟1
(1东北石油大学化学化工学院,黑龙江 大庆 163318;2中国石化胜利油田分公司地质科学研究院,山东 东营 257015)
2015-07-08收到初稿,2015-08-22收到修改稿。
联系人:丁伟。第一作者:单晨旭(1990—),女,硕士研究生。
摘要:采用分子动力学模拟(MD)的方法在分子层面上考察辛基酚聚氧乙烯醚磺酸盐(OPES)在油-水界面的界面行为。模拟结果表明:辛基酚聚氧乙烯醚磺酸盐可以大幅降低油-水界面的界面张力,在OPES浓度达到饱和浓度时,系统界面张力仅为3.85 mN·m-1;OPES中磺酸基是主要亲水基团,具有良好的亲水性;温度在318~373 K时,界面张力由24.63 mN·m-1下降到17.43 mN·m-1,这说明OPES具有良好的抗高温性能;当Na+浓度在1%~5%的环境下OPES性质稳定,界面张力仅有4.47 mN·m-1的小幅增加,因此OPES具有良好的耐盐性,并且其对Na+的耐盐性能好于对Ca2+的耐盐性。
关键词:辛基酚聚氧乙烯醚磺酸盐;分子动力学模拟;界面张力;抗温;抗盐
引 言
在三次采油中,为提高原油采收率,经常利用表面活性剂来降低油水界面张力,目前国内部分油田综合含水量已高达90%,单独的阴离子、非离子型表面活性剂已经不能满足当前的采油要求,阴非离子型表面活性剂作为一种同时有非离子及阴离子表面活性剂优点的两性表面活性剂对于目前日益严苛的采油环境的适应性更强[1-2]。本文研究的辛基酚聚氧乙烯醚磺酸盐(OPES)是一种具有优良的乳化、耐温、耐盐性能的阴非两性表面活性剂,它已经作为分散剂、润湿剂、乳化剂、洗涤剂等被广泛地应用于石油、日化、纺织等领域[3-5]。
分子动力学模拟主要是利用牛顿力学来模拟分子的运动,从不同状态下的体系抽取样本进行构型积分并以此为基础计算体系的热力学量等宏观性质。从20世纪90年代后期,人们开始利用计算机模拟研究表面活性剂的性能,它可以将真实环境中的实验现象在分子层面进行解释[6-10]。对液液界面的研究作为分子动力学模拟的重要研究方向之一近年来受到广泛的关注和报道,如Jang等[11]利用MD模拟了苯磺酸基在不同位置时十六烷基苯磺酸盐的界面张力等界面性能。Wardle等[12]考察了表面活性剂对无机盐、水和正己醇构成的混合物中钠离子迁移的影响。陈贻建等[13]用MD模拟方法对表面活性剂在气-液、固-液、液-液界面的自组装现象进行深刻解释分析。因此利用MD方法研究表面活性剂的界面张力、抗温、抗盐等界面性能具有重要意义。国内对于应用分子动力学模拟来研究表面活性剂性能的起步较晚,特别是对具有耐温、耐盐性能的表面活性剂的研究较少,本文通过分子动力学模拟来研究辛基酚聚氧乙烯醚磺酸盐的油-水界面行为、抗温、抗盐性能,可为实际实验提供较为准确的指导。
1 分子动力学模拟的模型选择与模拟方法
20世纪80年代以来,人们相继研发出可以适合不同环境的力场,如GROMOS、OPLS、AMBER、CHARMM等。本文选择Gromacs[14]中 GROMOS53a6[15-16]力场,以辛基酚聚氧乙烯醚磺酸盐为研究对象进行模型构建。
分子的物理化学性质由其分子结构决定,因此合理的分子结构以及准确的原子电荷是模拟准确性的基础保证。首先要对模拟对象用GAMESS(US)[17-18]进行结构优化,然后利用Kollman-Singh方法计算电荷,另外如果分子内存在对称结构还需进行电荷平均化来保证电荷分配的合理性。由于本文采用联合原子力场,因此还要去除sp3杂化。图1为优化后的OPES分子结构以及电荷分布,图中绿色小球为碳原子,白色小球为氢原子,红色小球为氧原子,黄色小球为硫原子。
在进行分子动力学模拟之前构建出合理的力场是极为重要的工作[19]。本文通过Autom-ated Topology Builder (ATB) and repository[20]生成的GROMOS[15]系列力场参数,利用现有的数据库以及量子化学进行计算,同时它可以充分考虑到分子中的对称结构,使其反映出的分子性质及参数更为精确。但ATB只能处理原子数小于40的分子,对于分子数大于40的分子结构需进行拆分。在获取准确的电荷及键参数之后利用packmol[21-22]程序定向排列分子将其堆砌成立方体结构。此外,本文选取的油-水界面需使表面活性剂平均分布在水相两侧,亲水基靠近水相,疏水基靠向油相。图2为初始状态下体系截图,其中中间红色部分为水分子,左右两侧蓝色部分为癸烷分子,油水中间即OPES分子。
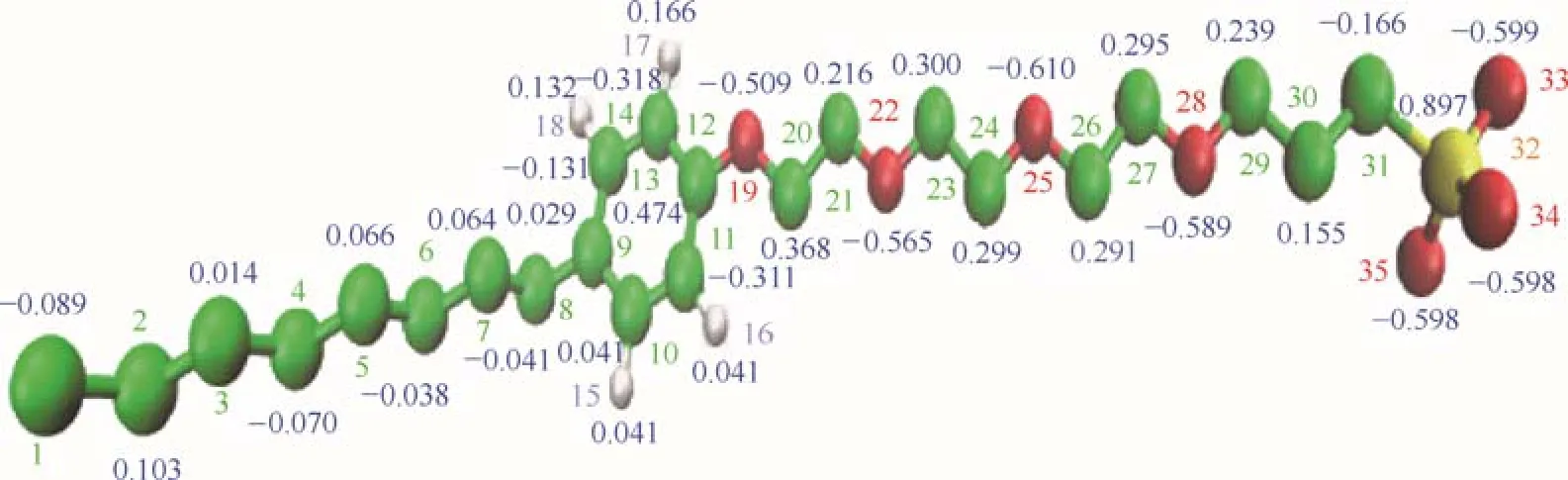
图1 辛基酚聚氧乙烯醚磺酸盐的分子结构以及电荷分布Fig.1 Molecular structures and united atom charge distribution of OPES
本文中所有体系所堆砌的盒子均为5 nm×5 nm×17.5 nm长方体,并在x、y、z方向选择周期性边界条件。系综选择NPT(等粒子等温等压系综),初始压力为1.01325×105Pa,水模型使用SPC[23](simple point charge),温度采用Nose-Hoover[24]热浴法,压力采用Parrinello-Rahman[25]压浴法,由于模拟过程中系统为等压变化所以本文模拟的所有系统最终压力值均在1.0081×105~1.0178×105Pa之间。在体系能量最小化后,先进行100 ps的NVT模拟,使体系升温到300K并在此温度下产生初速度,再进行1 ns的NPT模拟使体系密度达到合理状态,再进行12 ns的NPT模拟,控温及控压的弛豫时间为0.5、4.0 ps,积分步长为2 fs,在模拟过程中添加适当的阴阳离子保持体系为电中性。
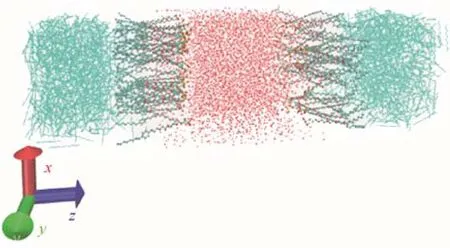
图2 初始状态下体系截图Fig.2 Snapshot of simulation system
2 模拟结果与分析
2.1界面性能
为考察OPES的界面行为及性能进行了8组对比模拟实验,分别记作S20、S50、S80、S100、S120、S140、S160、S180,即通过改变表面活性剂的数量对比各个体系的各相密度、界面宽度、界面张力以及界面聚集形态等界面行为,得出OPES表面活性剂浓度对界面行为的影响以及变化趋势等结论。
对比模拟实验均在由500个癸烷分子、5000个水分子构成的油-水界面以及温度为318 K的条件下进行。所有体系在平衡后表面活性剂的亲水基插入水相,亲油基插入油相并且形成非常稳定的界面。当表面活性剂浓度不断增加(从S20到S160)时由于单个表面活性剂分子的占有面积逐渐减少,分子的排列呈由分散变为紧凑的趋势。但当表面活性剂数量增大到180时,部分分子开始脱离原来平面,此时表面活性剂浓度已达到饱和状态。这一过程的界面张力变化如表1所示。
疏水尾链碳原子序参数(order parameter,SCD)可以用来表示疏水尾链的有序性
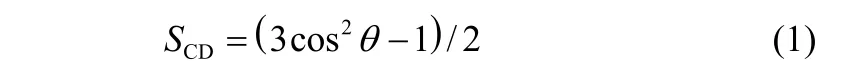
SCD可用式(1)来计算,θ代表Cn1-和Cn1+原子之间向量与界面垂直方向的角度。 图3所示为上述8个体系的序参数曲线,对于每一条序参数曲线都随着碳原子序号的增加序参数逐渐增大,这说明了疏水链末端的碳原子有序性更强。从图中还可以观察到S20的曲线几乎水平,这是由于OPES在界面的浓度过低其分子可以自由摆动。当表面活性剂的数量从20增加到180时,SCD曲线不断上升,这说明随着表面活性剂数量的增加疏水链排列的有序性也在不断增强。并且S160和S180的序参数曲线相当接近,这表明此时表面活性剂的浓度已经达到界面的饱和浓度。
对于表面活性剂来说降低界面张力的能力是考察其性能好坏的重要指标之一,下面通过考察不同体系的界面张力和界面宽度来进一步说明OPES的界面性能,如表1所示。对于界面张力可以利用式(2)来计算,其中LZ为盒子高度;Pxx、Pyy、Pzz分别为x、y、z方向的压力。
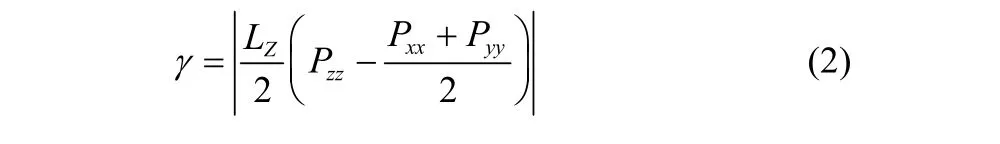

表1 不同表面活性剂浓度下体系界面张力和界面宽度Table 1 Interfacial tension and width of different surfactant concentration
从表1可以发现表面活性剂数量的增加使得界面张力逐渐下降。其中当OPES数量由20增加到80时,界面张力值较高,这说明当表面活性剂浓度较低时并不能起到很好的降低界面张力的作用;随着表面活性剂数量进一步增加界面张力逐渐下降,当OPES数量为180时,达到临界饱和状态,此时界面张力仅为3.85 mN·m-1,此变化规律与真实实验规律相同[4]。这同时也说明辛基酚聚氧乙烯醚磺酸盐可以很好地降低界面张力,是一种性能优良的表面活性剂。界面宽度用体系密度图中表面活性剂的密度曲线宽度来表示。随着表面活性剂个数增加界面宽度递增,起初界面宽度增加速度较快是因为界面OPES浓度过低并未饱和;当OPES数量达到100~140时,界面宽度仅有少量缓慢增加这是由于此时界面正在逐渐接近饱和状态。随着OPES的数量达到160和180时,界面宽度增加幅度变大,进一步验证此时界面已达到饱和状态。
本文提取体系稳定的S80、S120进行分析,两个体系的各部分密度图如图4所示。在平衡状态下两体系中水的平均密度分别为989.86 kg·m-3、992.43 kg·m-3,与国际温标318 K时水密度990.2 kg·m-3值接近;另外,两体系中癸烷平均密度为711.49 kg·m-3、710.72 kg·m-3,与真实状况下癸烷密度711.2 kg·m-3值接近,这表明模拟体系的模型选择、力场参数都是准确的,可以得出可靠结论。
在OPES结构中,有两个亲水基团分别为氧乙烯基(OG)、磺酸基(SDMSO),本文通过径向分布函数(通常指的是给定某个粒子的坐标,其他粒子在空间的分布概率)来对比两者与水之间的作用力。图5为表面活性剂中氧乙烯基和磺酸基与水分子中氢原子的径向分布函数、。由图可知,(r )曲线第1个峰值出现在0.306处,这表明磺酸基中的氧原子与水g中的氢原子之间较强的氢键作用形成了第1水层;在距离为0.458时,出现第2个峰值,数值有所下降,这表示逐渐减小的氢键作用形成了第2水层;第3水层形成在0.688处,此时磺酸基与水的作用进一步减弱,但3处的峰值均远大于氧乙烯基的峰值。这表明磺酸基与水分子的作用力远高于氧乙烯基,所以磺酸基为辛基酚聚氧乙烯醚磺酸盐结构中的主要亲水基团。
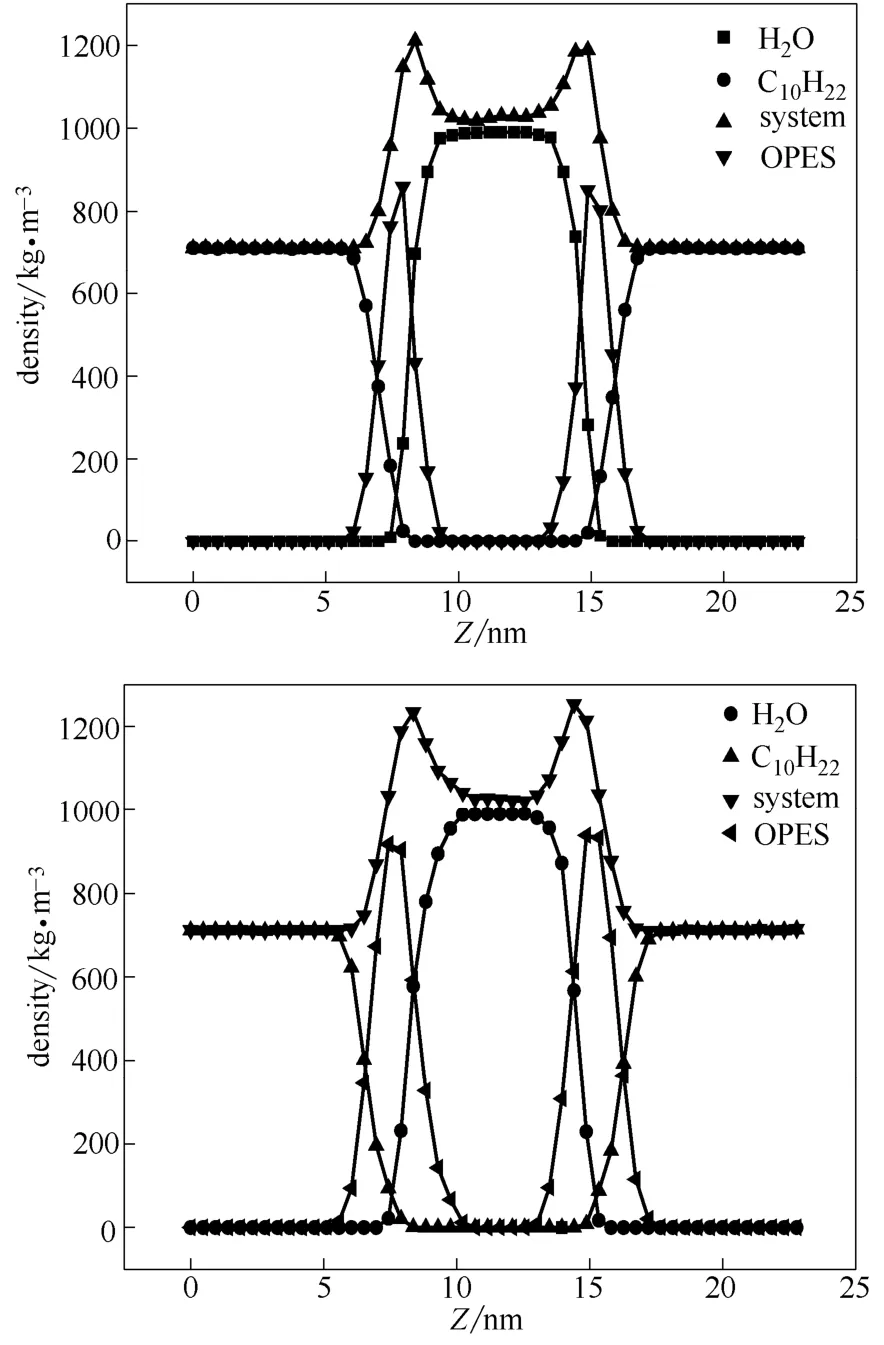
图4 S80、S120平衡状态下各部分密度分布Fig.4 Density profile of S80 and S120 simulation system under equilibrium state
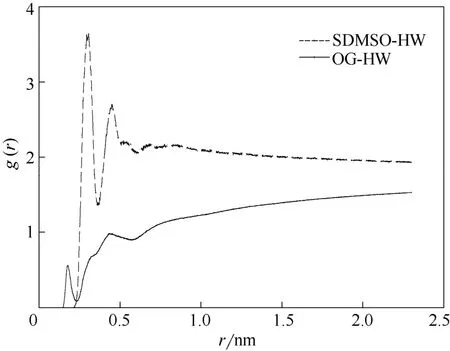
图5 S80中表面活性剂中亲水基团与水中氢原子之间的径向分布函数Fig.5 RDF between hydrophilic group and hydrogen of water molecule for S80 simulation system
2.由于采油环境日益严苛,一些表面活性剂在高温条件下与水之间的氢键易断裂,使得其亲水性能大幅降低,因此抗高温性能是考察表面活性剂好坏的重要指标之一。
本文选取4组对比模拟实验,保持表面活性剂数量为80不变,控制温度分别为318、343、358、373 K,记作S80T318、S80T343、S80T358、S80T373。
表2通过界面张力、表面活性剂与水的氢键数量、势能3组数据对比得出表面活性剂的界面张力随着温度升高而降低的结论。数据表明在4组模拟实验过程中,界面的宽度并没有发生改变。因此界面张力下降的主要原因是由于OPES势能的降低导致分子之间的作用力也随之降低。
2 温度对癸烷+水+OPES体系油-水界面张力的影响
另一个值得注意的改变是虽然随着温度的升高OPES与水之间的氢键有微量的下降但并没有达到浊点,况且磺酸基具有良好的亲水性,因此,OPES并没有因为温度升高而失效,反而能提高其在油-水界面的性能。
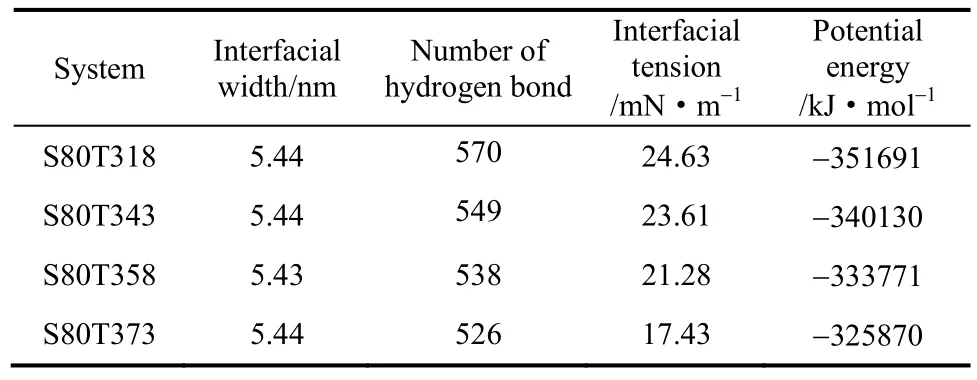
表2 不同温度下各体系的界面性能Table 2 Interfacial properties of systems at different temperatures
2.3盐对癸烷+水+OPES体系油-水界面张力的影响
大量数据表明,在高盐油藏表面活性剂的化学稳定性易受到影响,其结构可能受到改变或破坏进而影响石油采收率。石油磺酸盐、烷基苯磺酸盐等表面活性剂在高盐度的环境下极易失去活性[26-27]。因此,表面活性剂是否具有良好的抗盐性能显得尤为重要。对于辛基酚聚氧乙烯醚磺酸盐从结构上来说其具有的磺酸基结构应使其有良好的耐盐性能。
本文选取5组对比模拟实验,保持OPES数量为80、温度为318 K不变,分别向体系内加入1%、2%、3%、4%、5%的NaCl溶液,记作S80Na1、S80Na2、S80Na、S80Na4、S80Na5。图6所示为S80Na2体系在平衡状态下界面状态,其中蓝色小球为Na+。Na+几乎全部分散于水相中,在表面活性剂附近的分布很少,因此可以初步确定盐对表面活性剂的影响较小,辛基酚聚氧乙烯醚磺酸盐具有抗盐性。
为进一步确定OPES的耐盐性能,可以再通过不同体系平衡状态时相应的界面张力和氢键数量来讨论,相关数据如表3所示。
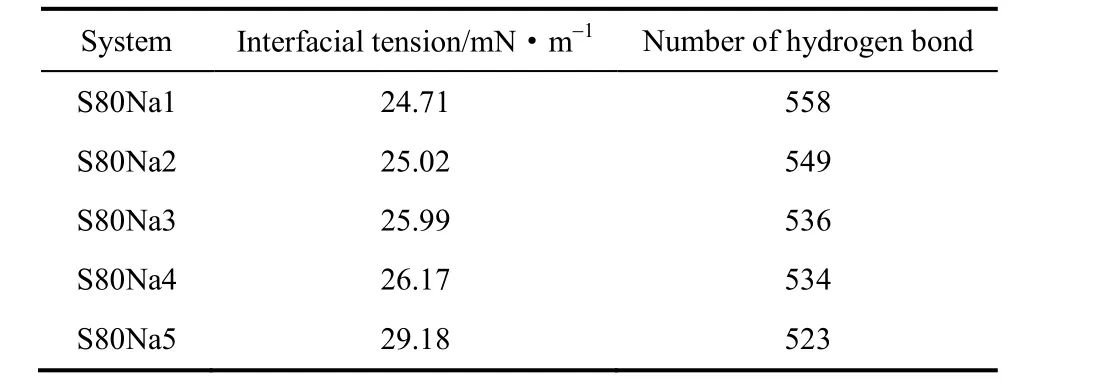
表3 不同浓度Na+溶液体系界面张力以及OPES与H2O的氢键数量Table 3 Number of hydrogen bonds between OPES and water and interfacial tension in systems with different concentrations of Na+
模拟数据显示,随着NaCl浓度的升高,表面活性剂在油水界面的界面张力仅有小幅升高,这是由于体系中不断加入NaCl使得OPES更加亲油,使得部分表面活性剂分子向油相中跃迁。另外,氢键数量有少量下降这是由于在体系不断添加Na+、Cl-过程中,替换了水相中的水分子使得水分子数量减少从而影响了氢键数量。
图7中显示了在不同NaCl浓度的体系中,磺酸基中的氧原子与水中氢原子的径向分布gOS-HW( r ),可以看出其峰值并没有因为NaCl浓度的增加而发生很大改变,这更能说明阳离子并不能对表面活性剂的性能造成影响。
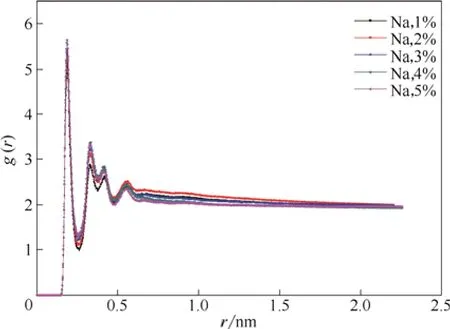
图7 在不同Na+浓度体系中磺酸基中的氧原子与水中氢原子的径向分布函数Fig.7 RDF between oxygen of sulfonate group and hydrogen of water molecule for different concentrations of Na+
下面同样通过疏水链碳原子序参数的变化来进一步验证OPES的抗盐性。从图8中可以看出,在同一体系中随着碳原子的增加序参数值增大,这说明越接近疏水尾链的末端的碳原子有序性越好。同时,对于NaCl浓度为1%、3%、4%、5%的体系,疏水尾链碳原子的序参数并未发生太大改变,NaCl浓度为2%时其序参数值还要大于1%时的序参数值,这说明在浓度为2%的NaCl溶液中疏水链碳原子间的相互作用力最强。
提取2%NaCl浓度时体系的疏水尾链碳原子序参数与同浓度的CaCl2体系进行对比,对比结果如图9所示。在CaCl2溶液中疏水链碳原子的SCD值明显高于无盐溶液以及2%的NaCl溶液中的SCD值,因此,在Ca2+的环境下碳原子的摆动空间与灵活性要小于在Na+的环境中。
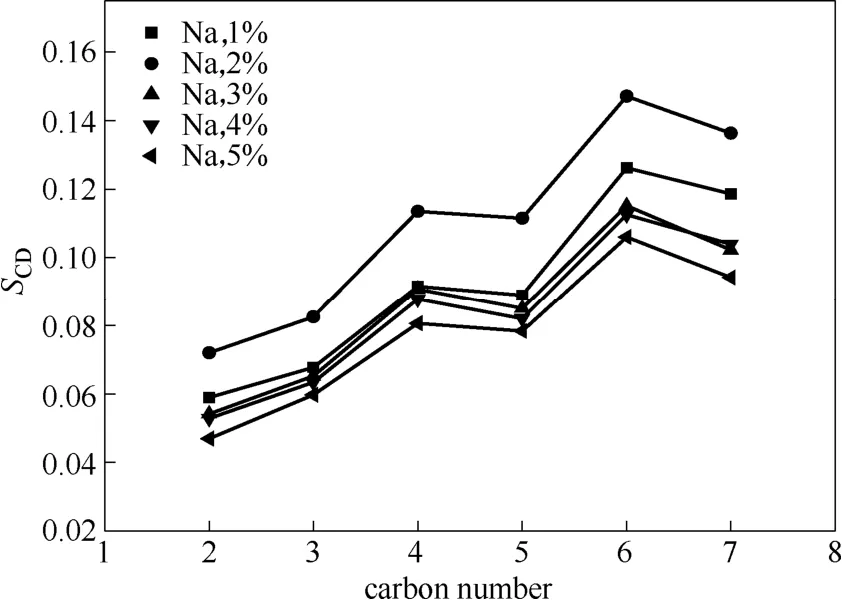
图8 不同Na+浓度下疏水尾链碳原子序参数Fig.8 Order parameter of hydrophobic chain atoms with different amount of Na+

图9 在不同离子溶液中疏水链碳原子的序参数Fig.9 Order parameter of hydrophobic chain atoms with Na+and Ca2+
图10中曲线分别代表在2%NaCl溶液、2%CaCl2溶液中磺酸基中的氧原子与水中氢原子之间的径向分布函数,如图所示两条曲线的峰值并未有太大差别,这说明OPES对Na+、Ca2+都有很好的抗盐性。
进一步分析OPES对Na+、Ca2+抗盐性的差别,考察了磺酸基中的氧原子与Na+、Ca2+的径向分布函数,如图11所示。图中两曲线的峰值出现较大差距,其中Na+曲线的峰值明显小于Ca2+曲线的峰值,这表明亲水基团与Na+的作用较小,也就是说Na+对OPES的性质影响较小。因此,辛基酚聚氧乙烯醚磺酸盐的抗盐性顺序为Na+>Ca2+,与对序参数所做的分析结论相同。
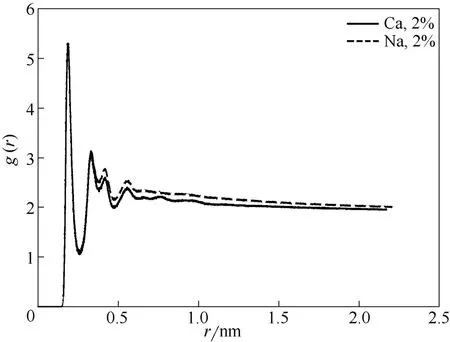
图10 在不同离子溶液中磺酸基中的氧原子与水中氢原子的径向分布函数Fig.10 RDF of interaction between oxygen of sulfonate group and hydrogen of water molecule for solutions with Na+and Ca2+
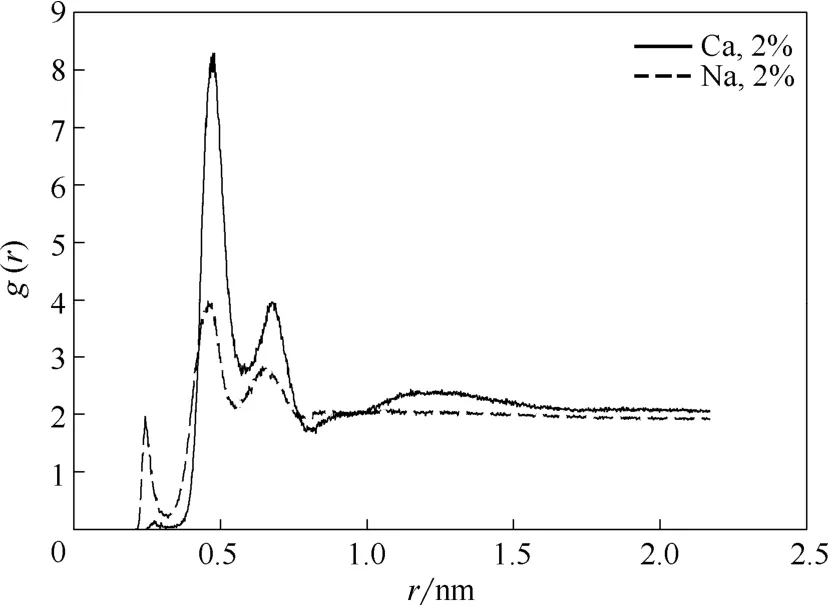
图11 磺酸基中的氧原子与不同离子之间的径向分布函数Fig.11 RDF of interaction between oxygen of sulfonate group and different ions
3 结 论
(1)分子动力学模拟可以准确模拟辛基酚聚氧乙烯醚磺酸盐在油-水界面的界面行为及性能。
(2)辛基酚聚氧乙烯醚磺酸盐可以大幅降低油-水界面的界面张力。
(3)辛基酚聚氧乙烯醚磺酸盐中磺酸基为其主要亲水基团且疏水链尾端碳原子有序性较好。
(4)辛基酚聚氧乙烯醚磺酸盐在温度为318~373 K时界面张力随温度升高而减小,具有良好的抗高温性能。
(5)辛基酚聚氧乙烯醚磺酸盐在Na+浓度为1%~5%的高盐条件下性质稳定,界面张力仅有小幅增加,并且其对Na+的耐盐性好于对Ca2+的耐盐性。
References
[1]韩巨岩,王文涛,崔昌亿,等. 烷基苯基聚氧乙烯醚磺酸盐 [J].日用化学工业,1998,28 (6): 60-62. DOI:10.13218/j.cnki.csdc.1998. 06.016. HAN J Y,WANG W T,CUI C Y,et al. Alkyl phenyl polyoxyethylene ether sulfonate [J]. China Surfactant Detergent & Cosmetics,1998,28 (6): 60-62. DOI:10.13218/j.cnki.csdc.1998.06.016.
[2]唐红娇,侯吉瑞,赵凤兰,等. 油田用非离子型及阴-非离子型表面活性剂的应用进展 [J]. 油田化学,2011,28 (1): 115-118. TANG H J,HOU J R,ZHAO F L,et al. Application progress of nonionic and anionic-nonionic surfactants used in oil field [J]. Oilfield Chemistry,2011,28 (1): 115-118.
[3]王业飞,李继勇,赵福麟. 高矿化度条件下应用的表面活性剂驱油体系 [J]. 油气地质与采收率,2001,8 (1): 1-7. DOI: 10.13673/j.cnki.cn37-1359/te.2001.01.019. WANG Y F,LI J Y,ZHAO F L. Surfactants oil displacement system in high salinity formations [J]. Oil & Gas Recovery Technology,2001,8 (1): 1-7. DOI :10.13673/j.cnki.cn37-1359/te.2001.01.019.
[4]贺伟东,李瑞冬,葛际江,等. 辛基酚聚氧乙烯醚磺酸盐的合成与界面张力的测定 [J]. 石油化工高等学校学报,2010,23 (4): 20-24. DOI:10.3696/j.issn.1006-396X.2010.04.005. HE W D,LI R D,GE J J,et al. The synthesis and interfacial tension determination of octylphenol polyoxyrethylene ether sulfonate [J]. Journal of Petrochemical Universities,2010,23 (4): 20-24. DOI: 10.3696/j.issn.1006-396X.2010.04.005.
[5]张永民,牛金平,李秋小. 壬基酚醚磺酸钠及其与重烷基苯磺酸钠复配物的耐盐性 [J]. 油田化学,2009,26 (1): 72-75. ZHANG Y M,NIU J P,LI Q X. The salt-tolerance of sodium nonyl phenol polyoxyethylene ether sulfonates and their mixture with a heavy alkylbenzene sulfonate [J]. Oilfield Chemistry,2009,26 (1): 72-75.
[6]王峰,艾池,曲广淼,等. 十二烷基磺丙基甜菜碱表面活性剂的气液界面聚集行为分子动力学模拟 [J]. 计算机与应用化学,2014,31 (7): 833-837.DOI:10.11719/com.app.chem20140715. WANG F,AI C,QU G M,et al. Molecular dynamics simulation on the aggregation behavior of N-dodecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate at air/water interface [J]. Computers and Applied Chemistry,2014,31 (7): 833-837. DOI:10.11719/com.app.chem20140715.
[7]李亚娉. 表面活性剂界面行为和构效关系研究 [D]. 济南: 山东大学,2014: 1-4. LI Y P. Interfacial behavior and structure-function relationship of surfactants [D]. Jinan: Shandong University,2014: 1-4.
[8]WANG L,HU Y H,SUN W,et al. Molecular dynamics simulation study of the interaction of mixed cationic/anionic surfactants with muscovite [J]. Applied Surface Science,2015,327 (327) 364-370.
[9]CHEN T,ZHANG G C,JIANG P,et al. Dilational rheology at air/water interface and molecular dynamics simulation research of hydroxyl sulfobetaine surfactant [J]. Journal of Dispersion Science & Technology,2014,35 (3): 448-455. DOI: 10.1080/01932691. 2013.785364.
[10]郑凤仙. 表面活性剂在固液界面及限制空间中的吸附和聚集行为的分子模拟研究 [D]. 北京: 北京化工大学,2009. ZHENG F X. Molecular simulation study on adsorption and aggregation behaviors of surfactants in a solid liquid interface and confined space [D]. Beijing: Beijing University of Chemical Technology,2009.
[11]Jang S S,LIN S T,MAITI P K,et al. Molecular dynamics study of a surfactant-mediated decane-water interface:effect of molecular architecture of alkyl benzene sulfonate [J]. The Journal of Physical Chemistry B,2004,108 (32): 12130-12140. DOI: 10.1021/jp048773n. [12]WARDLE K E,HENDERSON D J,ROWLEY R L. Molecular dynamics simulation of surfactant effects on ion transport through a liquid-liquid interface between partially miscible liquids [J]. Fluid Phase Equilibria,2005,233 (1): 96-102. DOI:10.1016/j.fluid. 2005.03.033.
[13]陈贻建,苑世领,徐桂英. 表面活性剂界面自组装的分子动力学模拟 [J]. 化学通报,2004,67 (11): 813-820. DOI:10.14159/j.cnki. 0441-3776.2004.11.005. CHEN Y J,YUAN S L,XU G Y. Molecular dynamics simulations for interfacial self-assembly of surfactant [J]. Chemistry,2004,67 (11): 813-820. DOI:10.14159/j.cnki.0441-3776.2004.11.005.
[14]丁伟,李思琦,宋晓伟. 基于分子动力学模拟的表面活性剂力场界面的构建及分析 [J]. 化学通报,2014,77 (10): 973-973. DING W,LI S Q,SONG X W. Building and analysis of interface of surfactant’s force field based on molecular dynamics simulation [J]. Chemistry,2014,77 (10): 973-973.
[15]OOSTENBRINK C,VILLA A,MARK A E,et al. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6 [J]. Journal of Computational Chemistry,2004,25 (13): 1656-1676. DOI: 10.1002/jcc.20090.
[16]CHRIS O,THEREZA A S,NICO F A,et al. Validation of the 53A6 GROMOS force field [J]. European Biophysics Journal,2005,34 (4): 273-284. DOI:10.1016/S0732-8893(01)00362-5.
[17]PERRA M J,WEBER S H. Web-based job submission interface for the GAMESS computational chemistry program [J]. Journal of Chemical Education,2014,91 (12): 2206-2208.
[18]SCHMIDT M W,BALDRIDGE K K,BOATZ J A,et al,General atomic and molecular electronic structure system [J]. Journal of Computational Chemistry,1993,11 (14)1347-1363. DOI:10.1002/ jcc.540141112.
[19]丁伟,高翔. 表面活性剂GROMOS53a6力场参数文件的构建 [J].精细石油化工进展,2013,14 (3): 1-4. DING W,GAO X. Method of generating GROMOS53a6 forcefield topology file for surfactant [J]. Advances in Fine Petrochemicals,2013,14 (3): 1-4.
[20]MALDE A K,ZOU L,BREEZE M,et al. An automated force fieldtopology builder(ATB) and repository: version 1.0 [J]. American Chemical Society,2011,7 (12): 4062-4037. DOI:10.1021/ct200196m.
[21]MARTĺNEZ L,ANDRADE R,BIRGIN E G,et al. PACKMOL:a package for building initial configurations for molecular dynamics simulations. [J].Journal of Computational Chemistry,2009,30 (13): 2157-2164. DOI:10.1002/jcc.21224.
[22]MARTĺNEZ J M,MARTĺNEZ L. Packing optimization for automated generation of complex system’s initial configurations for molecular dynamics and docking [J]. Journal of Computational Chemistry,2003,24 (7): 819-825. DOI: 10.1002/jcc.10216.
[23]LOMBARDERO M,MARTLN C,Jorge S,et al. An integral equation study of a simple point charge model of water [J]. Journal of Chemical Physics,1999,110 (2): 1148-1153. DOI:10.1063/1.478156.
[24]TUCKERMAN M E,MARTYNA G J. Comment on simple reversible molecular dynamics algorithms for Nose-Hoover chain dynamics [J]. Journal of Chemical Physics,1999,111 (7): 3313.
[25]MANFRED H U. Comments on a continuum-related Parrinello-Rahman molecular dynamics formulation [J]. Journal of Elasticity,2013,113 (1): 93-112. DOI: 10.1007/s10659-012-9412-3.
[26]王翀,王瑞,丁伟,等. 2,5-二甲基十四烷基苯磺酸钠异构体的合成及其EACN值的测定 [J]. 精细石油化工,2009,26 (9): 8-11. WANG C,WANG R,DING W,et al. Synthesis of isomeric sodium 2,5-dimethylte tradecyl benzene sulfonates and determination of the EACN value [J]. Speciality Petrochemicals,2009,26 (9): 8-11.
[27]丁伟,宿雅彬,张春辉,等. 支链异构十五烷基间二甲苯磺酸钠溶液表面性质及其溶剂行为 [J]. 应用化学,2009,26 (9): 1023-1026. DING W,SU Y B,ZHANG C H,et al. Surface properties of branch in isomers of pentadecylm-xylene sulfonates solution and solvent behavior [J]. Chinese Journal of Applied Chemistry,2009,26 (9): 1023-1026.
Molecular dynamics simulation for interface behavior of octylphenol polyoxyethylene ether sulfonate
SHAN Chenxu1,CAO Xulong2,ZHU Yangwen2,LIU Kun1,QU Guangmiao1,LÜ Pengfei1,
XUE Chunlong1,DING Wei1
(1College of Chemistry and Chemical Engineering,Northeast Petroleum University,Daqing 163318,Heilongjiang,China;2Gelogical Scientific Research Institute,Shengli Oilfield Branch Company,Dongying 257015,Shandong,China)
Abstract:Behaviors of octylphenol polyoxyethylene ether sulfonate (OPES) molecules on the oil-water interface were studied through molecular dynamics simulation (MD). The results showed that OPES could weaken tension of the oil-water interface significantly. The interface tension was only 3.85 mN·m-1at OPES saturation. The sulfonic group in OPES was the main hydrophilic group and had good hydrophilcity. The interface tension declined from 24.63 mN·m-1to 17.43 mN·m-1when temperature increased from 318 K to 373 K,indicating the good high temperature resistance of OPES. OPES maintained the stable properties within 1%—5% Na+concentration with only 4.47 mN·m-1increase of the interface tension. Therefore,OPES had good salt tolerance and could tolerate higher Na+concentration than Ca2+.
Key words:octylphenol polyoxyethylene ether sulfonate; molecular dynamics simulation; interfacial tension; heat resistance; salt tolerance
基金项目:十二五国家重大专项(2011ZX05011-004)。
Received date: 2015-07-08.
Corresponding author:DING Wei,dingwei40@126.com
DOI:10.11949/j.issn.0438-1157.20151092
中图分类号:O 641
文献标志码:A
文章编号:0438—1157(2016)04—1416—08
