HCV NS3/4A蛋白酶与Faldaprevir类似物的分子识别机制及其复合物运动模式
2016-07-04梁立胡建平杜文义左柯刘嵬苟小军
梁立,胡建平,2,杜文义,左柯,刘嵬,苟小军
1 成都大学 四川抗菌素工业研究所 药食同源植物资源开发四川省高校重点实验室,四川 成都 6101062 乐山师范学院 化学学院,四川 乐山 614004
HCV NS3/4A蛋白酶与Faldaprevir类似物的分子识别机制及其复合物运动模式
梁立1,胡建平1,2,杜文义1,左柯1,刘嵬1,苟小军1
1 成都大学 四川抗菌素工业研究所 药食同源植物资源开发四川省高校重点实验室,四川 成都 610106
2 乐山师范学院 化学学院,四川 乐山 614004
梁立, 胡建平, 杜文义, 等. HCV NS3/4A蛋白酶与Faldaprevir类似物的分子识别机制及其复合物运动模式. 生物工程学报, 2016, 32(5): 669–682.
Liang L, Hu JP, Du WY, et al. Molecular recognition mechanism and motion of HCV NS3/4A protease with Faldaprevir analogue. Chin J Biotech, 2016, 32(5): 669–682.
摘 要:Faldaprevir类似物 (Faldaprevir analogue molecule, FAM) 能有效抑制HCV NS3/4A蛋白酶的催化活性,是一种潜在抗HCV先导化合物。通过生物信息学统计分析了已报道的HCV NS3/4A 蛋白酶晶体结构,得到了FAM-HCV NS3/4A蛋白酶晶体结构。对FAM-HCV NS3/4A蛋白酶复合物进行了20.4 ns的分子动力学模拟,重点从氢键和结合自由能两个角度分析了二者分子识别中的关键残基及结合驱动力。氢键和范德华力是促使FAM特异性结合到蛋白V132-S139、F154-D168、D79-D81和V55的活性口袋中的主要驱动力,这与实验数据较为吻合。耐药性突变实验分析了R155K、D168E/V和V170T定点突变对FAM分子识别的影响,为可能存在的FAM耐药性提供了分子依据。最后,用自由能曲面和构象聚类两个方法探讨了体系的构象变化,给出体系的4种优势构象,为后续的基于HCV NS3/4A蛋白酶结构的Faldaprevir类似物抑制剂分子设计提供一定的理论帮助。
关键词:HCV NS3/4A蛋白酶,Faldaprevir类似物分子,分子动力学模拟,结合自由能,构象变化
Received: August 31, 2015; Accepted: March 16, 2016
Supported by: National Natural Science Foundation of China (Nos. 11247018, 11147175), Research Key Project of Sichuan Province Education Department (No. 12ZA066), Leshan Science and Technology Project (No. 14SZD018).
Xiaojun Gou. Tel: +86-28-84616301; E-mail: gxjmeprd@163.com
国家自然科学基金 (Nos. 11247018, 11147175), 四川省教育厅科研重点项目 (No. 12ZA066), 乐山市科技计划项目 (No. 14SZD018)资助。
丙型肝炎病毒 (Hepatitis C virus,HCV) 感染是造成慢性肝病和肝移植的主要原因之一[1]。据估计,全球范围内有1.6-1.8亿人长期感染HCV[2-3],其中一部分会发展为进行性肝纤维化,最终导致肝硬化,晚期肝病及肝癌[4]。目前的丙型肝炎标准诊疗方案 (Standard of care,SOC) 是联合使用长效干扰素 (PEG-IFNα) 和广谱抗病毒药物利巴韦林 (Ribavirin)[5]。但这种治疗方案的持续病毒学应答 (Sustained viral responses,SVR) 受治疗周期、HCV基因型等因素的影响,在40%-80%大范围内波动[6],治疗效果往往不够理想。Pawlotsky课题组的研究结果表明[7],在SOC中加入直接作用性抗病毒(Directly acting antiviral,DAA) 药物后SVR显著提高,疗效令人满意。
HCV NS3/4A蛋白酶 (Protease,PR) 是由NS3丝氨酸蛋白与辅助因子NS4A蛋白以非共价键结合而成的二聚体。NS3丝氨酸PR是HCV非结构蛋白加工成熟过程的关键酶,与三磷酸核苷酸酶、RNA解螺旋酶结合,构成HCV复制所必需的非结构蛋白NS3。NS3属于胰PR超家族,拥有PR和RNA解螺旋酶双重活性[8],位于由HCV RNA编码的前体多聚蛋白的开放阅读框 (Open reading frame,ORF) 中的非结构蛋白区中。另外,NS3/4A PR能水解TRIF和MAVS两种人类免疫蛋白,从而干扰人体对HCV的先天性免疫应答[9-10]。NS3/4A PR还可通过调控IFN-3的表达水平,破坏细胞内免疫通路,诱导宿主产生免疫逃逸[11]。
HCV NS3/4A PR抑制剂是首类进入临床试验的DAA药物。Faldaprevir类似物分子 (Faldaprevir analogue molecule,FAM)[12]作为一种HCV NS3/4A PR抑制剂,能有效抑制NS3/4A PR活性。阻断HCV的复制、翻译和翻译后多聚蛋白的加工成熟,还可使IFN更好地发挥抗病毒疗效。
构象变化和分子识别是药物设计的基础。目前有关HCV NS3/4A PR与FAM之间识别机制和复合物体系在水中构象变化等问题尚未见报道。本文用分子动力学 (Molecular dynamics,MD) 模拟方法对这些问题进行了研究,为后续的结构改造、新药研发提供结构及理论支撑。图1给出了FAM的化学结构,图中数字表示原子序号,括号里的数字用来表示极性氢,这些极性氢有可能和蛋白质部分形成氢键[12]。
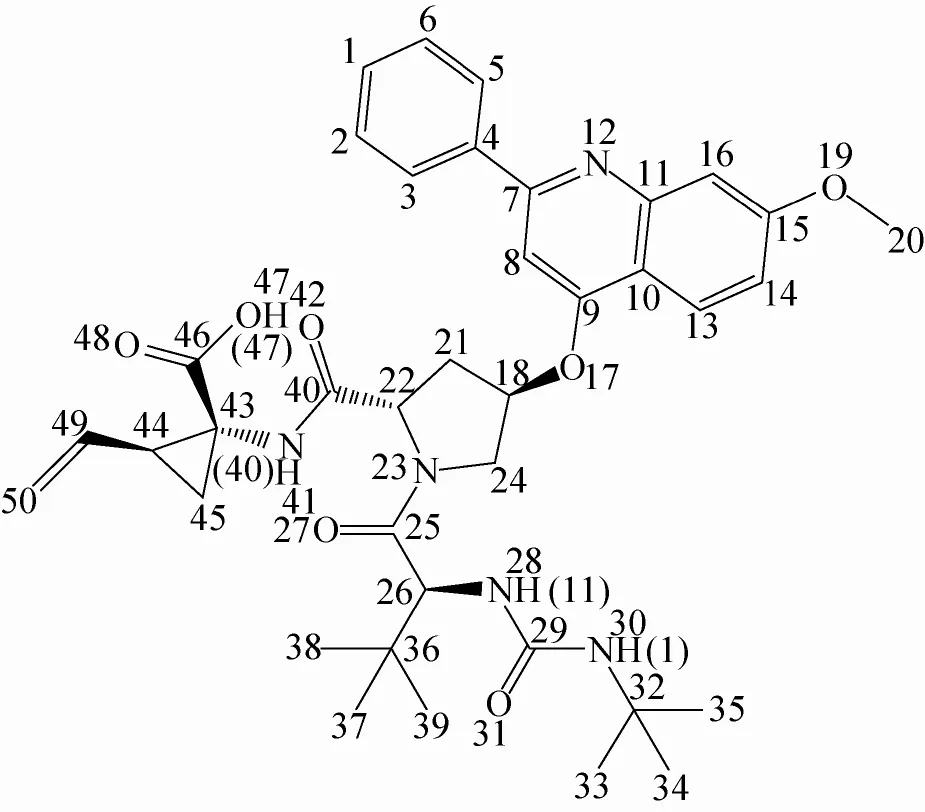
图1 FAM的分子结构[12]Fig. 1 Molecular structure of FAM[12].
1 模拟方法
1.1分子动力学模拟
在HCV NS3/4A PR和FAM的晶体结构[12](PDB代码:4K8B) 中,包含有辅助结晶的离子和结晶水,考虑到这些水分子等并未与FAM结合,也未参与活性中心的构成。故删去4条链的非蛋白部分,包括A链的ZN201、SO4203 和SO4204,B链的ZN201、SO4203,以及D链的SO4301及所有的结晶水分子。仅保留体系中PR的氨基酸与FAM部分,并以此作为MD模拟的初始结构。MD模拟采用AMBER[13]程序以及AMBER力场,其中生物大分子的力场参数基于实验值拟合[14]。模拟温度为26.85 ℃,溶剂使用TIP3P水模型[15]。
首先,将复合物置于去头八面体水盒子中,使复合物边缘距盒子壁0.8 nm,此时盒子里共加入9 820个水分子,体系总原子数为15 614。随后进行两次能量优化,第一次是约束溶质的10 000步能量优化,约束力常数为2.091×105kJ/(mol·nm2),由5 000步最陡下降法优化和5 000步共轭梯度能量优化组成。第二次是去约束的5 000步最陡下降和5 000步共轭梯度能量优化,收敛条件为能量梯度小于4.182×10-4kJ/(mol·nm2)。能量优化完成后,开始进行MD模拟,整个模拟过程分为两步分进行:首先进行约束动力学模拟,约束力常数为4.182×103kJ/(mol·nm2),期间温度使用Langevin dynamics升温机制,温度偶联因子设为1.0,温度从–273.15 ℃逐步升高到26.85 ℃,模拟400 ps;接着,再进行20 ns的无约束恒温MD模拟。用SHAKE算法[16]约束键长,非键半径为1 nm;模拟积分步长为2 fs,每隔1 ps采集一次构象,共采集20 400个构象。并使用VMD软件对体系模拟全过程跟踪监测。
1.2能量分解
在本文中,用MM/GBSA (Molecular mechanics/generalized Born surface area) 方法[17-18]进行能量分解分析。其基本思想是把每个残基的能量贡献近似分为力学(Molecule mechanics,MM) 方法计算的真空分子内能、用广义波恩 (Generalized Born,GB) 模型[19-20]计算的极性溶剂化能,以及用LCPO算法[21-22]计算的非极性溶剂化能,并且把能量分解到残基的主链原子核侧链原子上。这里,真空分子内能可分为极性的静电相互作用和非极性的范德华相互作用两部分。LCPO算法认为非极性溶剂化能与溶剂可接近表面积 (Solvent accessible surface area,SASA) 呈正相关。通过MM/GBSA能量分解,可以观察到HCV NS3/4A PR中的主要残基对FAM结合所做出的贡献。
1.3自由能曲面
自由能曲面 (Free energy landscape,FEL)是通过研究自由能曲面图的最小值 (即体系最稳定的状态) 和分界点 (即体系变化过程中的一个短暂的状态) 来表示生物学过程中发生的动力学变化的一种分析方法[23-26]。一些主成分(Principal component,PC) 可以用以表示重要的动力学过程,这些PC也可以来绘制自由能曲面图[27],具体根据公式:
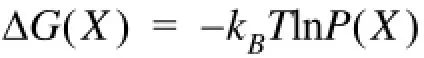
式中,坐标X表示PC,kB表示玻尔兹曼常数,T是绝对温度,P(X)是体系PC的概率。
1.4聚类分析
使用MMTSB工具[28],对MD模拟得到的20 400个体系构象进行聚类分析 (Cluster analysis)。分析过程依据Daura等[29]提出的观点:逐一计算各构象之间的RMSD值,以此为基础建立RMSD矩阵 (N×N, N为构象数)。人为设定一个RMSD阈值,在任意两构象RMSD值之间进行比较,若其差值小于此阈值则归为一类。以每一类中能量最低的构象作为该类的代表构象。本文中,以体系整体RMSD值建立矩阵,对体系整体构象的聚类情况进行了讨论。
2 结果与讨论
2.1HCV NS3/4A PR的生物信息学分析
截止目前,在蛋白质数据库 (www.rcsb.org)中检索HCV NS3/4A PR,得到相关结构104个。其中仅来源于丙型肝炎病毒的结构有81个,而其中结合了NS4A 蛋白的成熟复合物结构仅有17个。表1给出了蛋白质数据库中收录的成熟HCV NS3/4A PR结构生物学信息。表中PDB表示解析结构的PDB代码,Res.表示解析的分辨率,Sequence为HCV NS3/4A PR的氨基酸序列,ligand则表示结构中的小分子结合信息。
从表1可以看出,该17个结构分辨率相差较大,但均在可接受范围内。除4U01外均为二聚体结构,其中3LOX、3LON、3OYP、2XCN、2OBO、2A4Q、2F9U和1RTL的NS3A、NS4A蛋白单体氨基酸序列之间不一致,表中仅给出了相对较全的单体序列。考虑到后续对二聚体结构整体的模拟工作,避免二聚体结构不完整对模拟结果可能造成的误差,故没有选取这些结构。加之结合有FAM的晶体结构仅有4K8B一个,故选其用于后续的MD模拟。
2.2体系整体的模拟分析
图2显示了HCV NS3/4A PR和FAM的复合物体系在模拟过程中的势能随时间的变化。分子势能在400 ps以后达到平衡。经计算,平衡后的势能平均值为–4.26×105kJ/mol,标准偏差为2 000 kJ/mol,波动范围小于0.47%。由于模拟体系较大,共含有15 614个原子,波动对结果的影响不大,可以忽略。可见,HCV NS3/4A PR和FAM的复合物结构在模拟过程中维持稳定可靠。
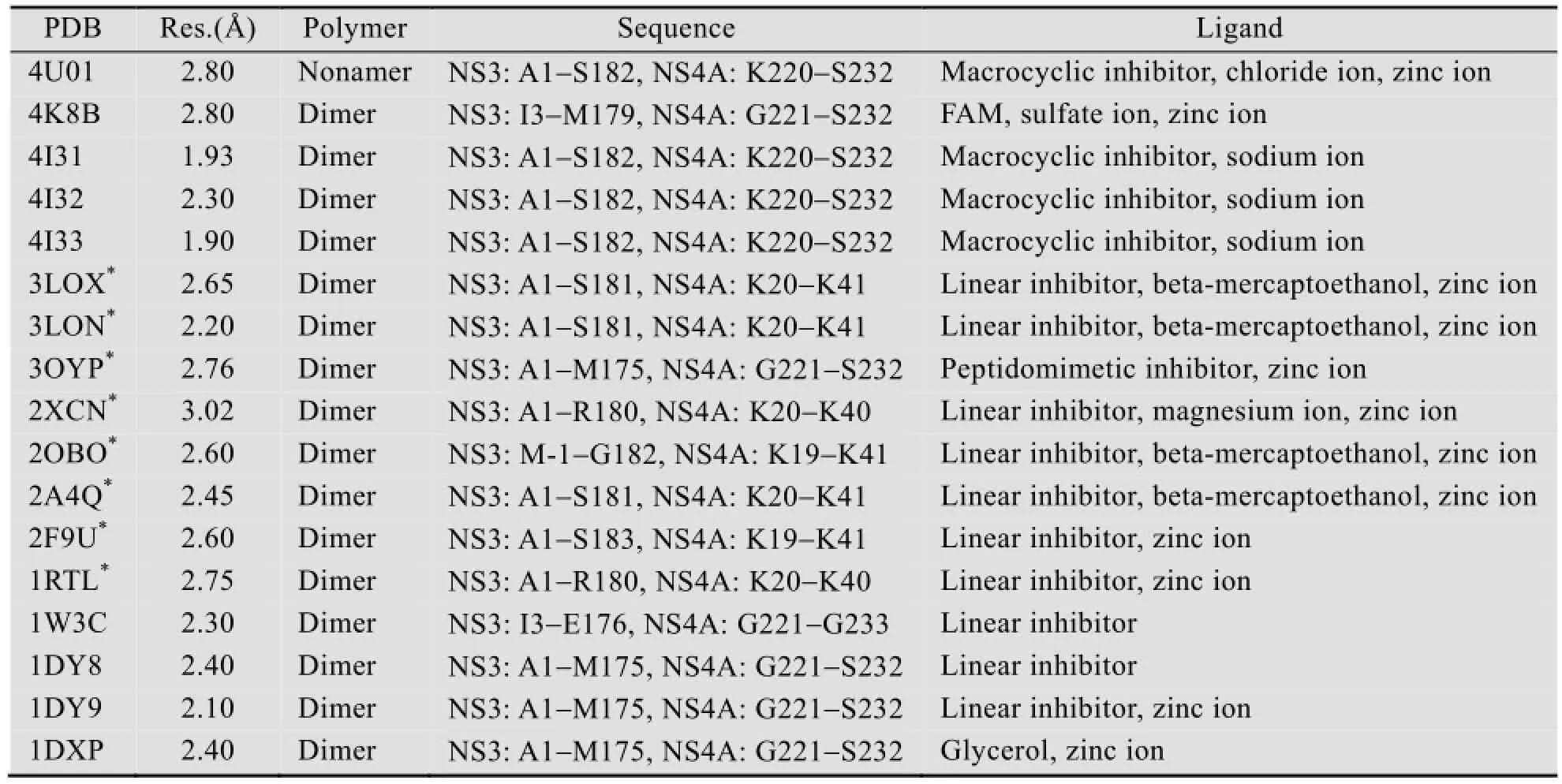
表1 HCV NS3/4A PR结构生物学信息Table 1 The structural biological information of HCV NS3/4A PR
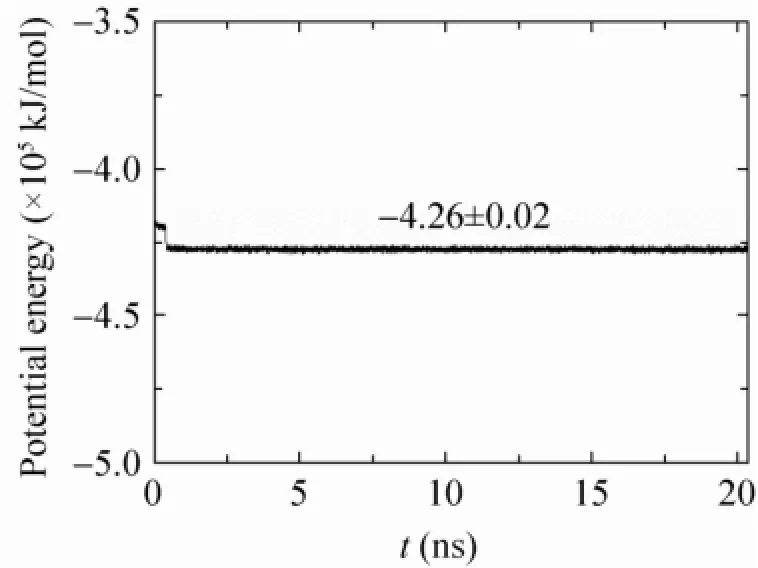
图2 体系势能随时间的变化Fig. 2 Potential energy of the system versus simulation time.
图3A给出了PR分子中A链、B链、C链和D链主链Cα原子的方均根偏差 (Root mean square deviation,RMSD) 随时间的变化。从图3A可以看出,蛋白酶在400 ps后基本趋于平稳。A、B链的RMSD高于C、D链。经计算,A链RMSD为 (0.125±0.020) nm、B链RMSD为(0.117±0.017) nm、C链和D链分别为(0.062±0.019) nm和 (0.060±0.017) nm。说明C、D链较A、B链稳定。这与其结构有关,A、B链较长,含177个氨基酸残基,为蛋白酶的主体部分,大部分暴露在溶剂中,更易受到溶剂的影响;而C、D链仅含12个氨基酸,分别结合与A、B链外侧的结合口袋中,受到口袋内氨基酸残基的牵制作用,稳定性较好。
图3B给出了体系整体主链Cα原子、配体FAM的ligand分子的RMSD随时间的变化。与图3A对比发现两个小分子的RMSD值在模拟过程中基本趋于平衡,经计算,ligand的RMSD 为 (0.173±0.024) nm,无较大波动。表明FAM稳定地结合在活性口袋中。而体系整体的RMSD数值则维持在较高位置,这可能是由于复合物分子较大,通过两分子FAM相互作用,形成二聚体结构,故在模拟过程中分子内有着较为显著的相对运动,4条链的相对位置及角度有较大变化。
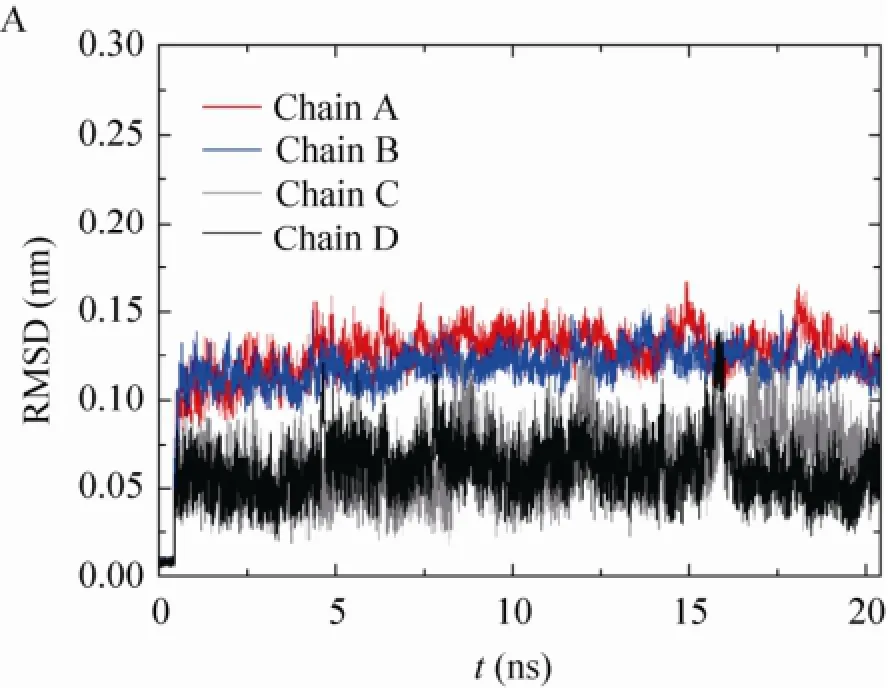
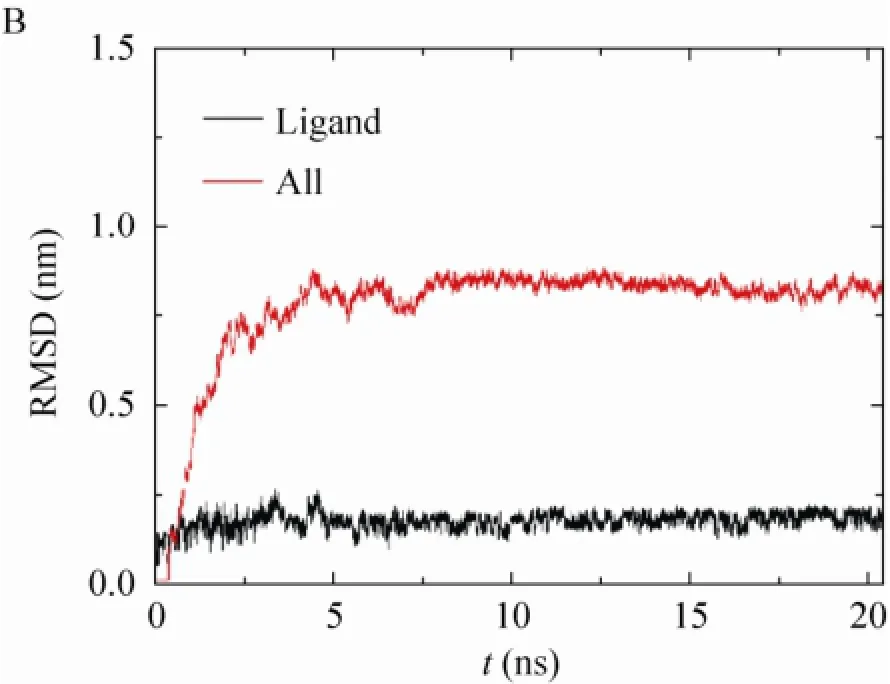
图3 蛋白质体系中Cα原子与FAM方均根偏差随时间的变化Fig. 3 RMSD of the Cαatoms in the proteinic system and FAM versus simulation time.
图4给出了复合物分子中蛋白酶B链和D链的主链Cα方均根涨落 (Root mean square fluctuation,RMSF) 分布。由于体系为二聚体结构,为简化分析运算,此处仅分析B链和D链。从图4中可以看到4个区域的涨落较大,分别是P96-S101、P115-R119、R123-L126、V158-V163。通过VMD观察这4个区域在水中模拟的运动轨迹,发现P96-S101段和P115-R119段是两个loop区,暴露在水中,与周围的残基作用较弱,所以柔性较大;R123-L126段和V158-V163段则是两个β折叠片,均与FAM结合部位相近。较大的运动幅度可能是受到了小分子的影响。D链仅有较小的涨落出现,说明其柔性较小。
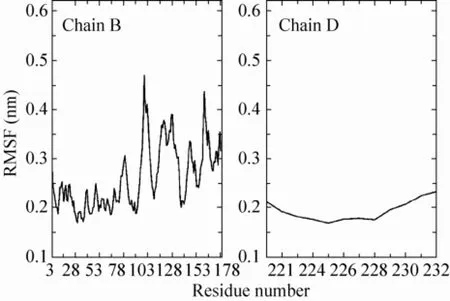
图4 体系B链和D链Cα原子的方均根涨落分布Fig. 4 RMSF distribution of the Cα atoms in chain B and chain D.
图5A给出了晶体结构中蛋白酶A链和B链温度因子 (B-factor,B因子) 的自相关性。可以看到,A链和B链的相关性很高,其相关系数为0.81,这表明模拟分析方法可靠。图5B给出了模拟计算得到的B因子与X-ray实验所得B因子的相关性。一般来说,体系较大的RMSF和B因子均可用于说明相应的残基部分柔性较大。经计算,二者相关性为0.68。考虑到样本数达到了177个,两组数据存在着较为显著的相关性,进而证明用模拟计算分析得到的柔性分布结论较为可靠。
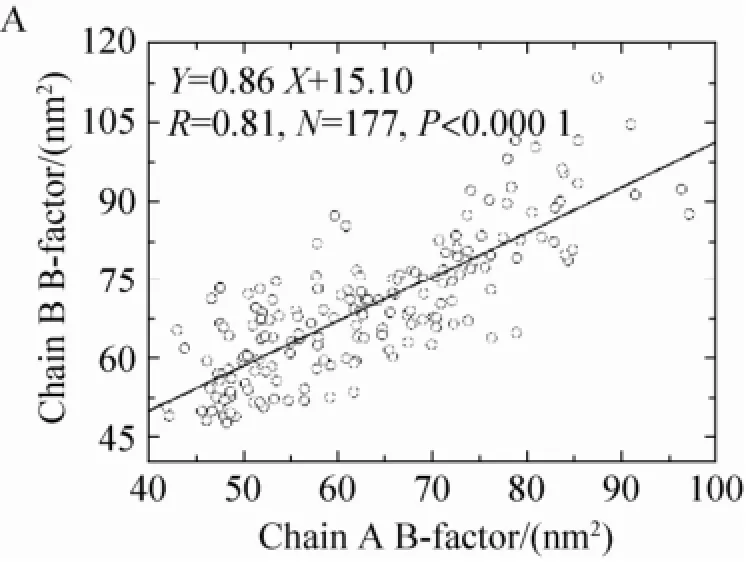
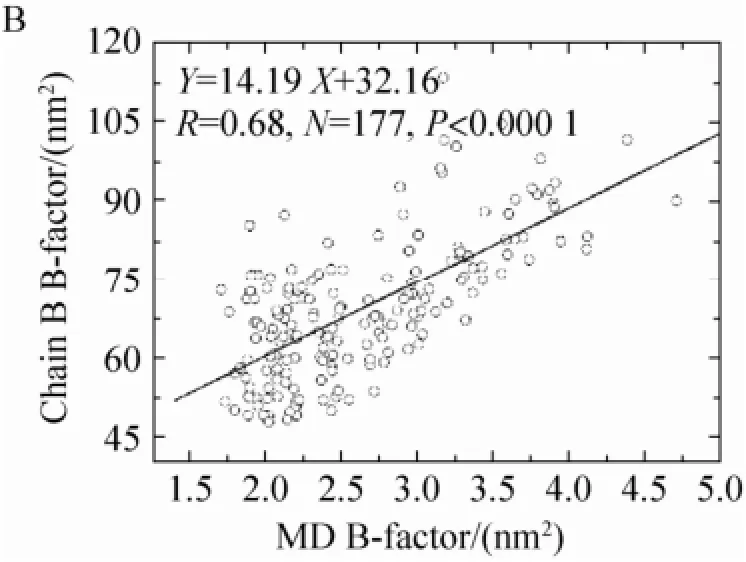
图5 晶体结构A链和B链的B因子相关性以及晶体结构B因子与分子模拟B因子数据的相关性Fig. 5 The correlation of B-factors for crystal structure from PDB of chain A with chain B, and that of calculated B-factor values in chain B from MD simulation with B-factors from crystal structure.
2.3分子识别
表2列出了各能量项对FAM与HCV NS3/4A PR结合自由能的贡献。结合自由能(Binding free energy) 对药物分子的活性评价起着重要的作用。表2中ELEIN表示真空状态下分子内能的静电能部分;VDWIN表示真空中分子内能的范德华能部分;ELEPB是指通过PB算法计算出的体系与溶剂之间的静电能;VDWPB是指通过PB算法计算得到的体系与溶剂之间的范德华能。Protease表示对HCV NS3/4A PR单独计算的能量结果;FAM则表示单独考虑FAM的能量结果;Complex表示对复合物进行模拟计算的能量值;Delta值则是复合物形成前后的能量变化。
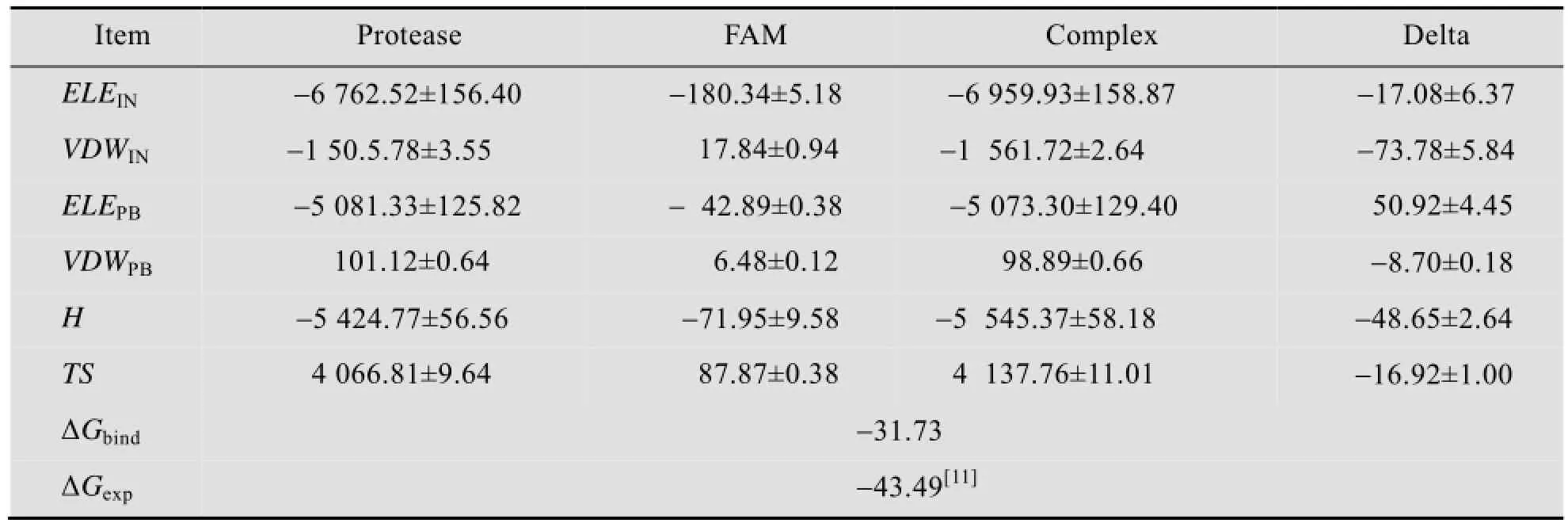
表2 各能量对结合自由能的贡献 (kJ/mol)Table 2 The contribution to binding free energy by each kind of energy (kJ/mol)
从表2可以看出,当FAM与PR结合成复合物时,体系内能减小。这一过程主要通过两条途径实现:一是体系内能的静电部分减小,释放了17.08 kJ/mol;二是体系内能的范德华部分的减小,释放了73.78 kJ/mol,促进了复合物的形成。体系溶剂化能的范德华部分也降低(-8.70 kJ/mol);但代替牺牲溶剂化能的静电部分在结合过程中则大幅增大 (50.92 kJ/mol),逆转了内能静电部分降低引起的促结合结果。
综上,非极性相互作用为FAM与PR识别的主要驱动力。此外,体系总焓变为-48.65 kJ/mol,总熵变为-16.92 kJ/mol,求得体系绝对结合自由能为-31.73 kJ/mol。实验文献[12]提到,与FAM结构类型很高的化合物抑制剂的实验结合自由能值ΔGexp。此化合物仅以O取代了N30,所以其实验数据有较高的参考价值。经对比,计算得出的理论值与实验值基本吻合。
氢键的存在可以维持复合物分子的稳定性,同时也可以增强受体-配体分子之间的特异性识别。为进一步探讨HCV NS3/4A PR与FAM的识别机制,表3列出了二者之间形成的氢键。氢键采用几何判据[30],供体原子-氢原子-受体原子之间的夹角大于135°,供体原子-受体原子之间的距离小于0.35 nm。表中ligand1和ligand2分别表示结合在A链和B链的FAM,Freq表示氢键占有率,即氢键在整个模拟过程中出现的概率,Freq越大表示对应的氢键越稳定。
从表3可以看出,FAM与A157、R155和S139形成了稳定的氢键,与X-ray实验数据[12]一致。其中,S139位于HCV NS3/4A PR的催化中心,是酶发挥生物活性的关键残基之一;R155和A157位于的扭曲片段也是PR抑制剂的重要结合部位[1]。FAM与PR之间较为稳定的3个氢键为维持复合物的稳定性,并有助于促进抑制剂的特异性识别,发挥较高的抑制活性 (Ki=0.02 μmol/L)。
表4列出了HCV NS3/4A PR与底物FAM识别的关键残基能量分解结果。考虑到体系为同源二聚体结构,并结合两分子FAM,为方便分析,此处仅研究了与B链结合的ligand2。表中的Resid表示关键残基的残基号,ligand1表示与A链结合的FAM;Evdw为真空范德华结合能;Eele为真空静电结合能;Egb为极性溶剂化结合能,Egbsur表示非极性溶剂化结合能。能量分解显示,小分子ligand1在结合过程中起着重要作用,说明复合物中FAM以双分子形式存在是其发挥药效的重要形式。B链的F154’-A157’段和R130’ -Y134’段也在结合中起着关键的作用,与有关文献[1]报道的HCV NS3/4A蛋白酶抑制剂结合部位基本一致。此外,V55’也在FAM的结合中作出了较大的贡献。Welsch等[31]曾探究过V55突变后产生酰胺基抵抗的现象,可见,V55’是FAM发挥抑制作用的关键残基。由此可以断定:HCV NS3/4A蛋白酶的F154-A157、R130-Y134为FAM结合的主要区域,V55与另一分子的FAM为维持复合物结构稳定,发挥抑制作用提供了重要保证。

表3 FAM和HCV NS3/4A PR之间所形成的氢键Table 3 The hydrogen bonds between FAM and HCV NS3/4A protease
图6A给出了在势能最低构象中,以ligand2 (FAM) 为中心,半径为0.4 nm范围内的接触残基。图中,D79’–D81’、V132’–S139’、F154’–D168’、用黄色solid-ribbon模式表示,A链的关键残基用红色stick模式表示,B链的关键残基用蓝色stick模式表示,FAM ligand1用 line模式表示。ligand2 (FAM) 用stick模式表示。从图中可以看出位于酶催化中心的H57’的咪唑环与FAM的喹啉环通过π-π作用结合在一起。表明蛋白酶此处活性口袋平坦,FAM的喹啉环为使其在活性口袋中结合地更加紧密,有利于提高抑制活性。图6B主要展示了FAM在复合物中的双分子结合模式。从图中可以看出,ligand2分子的苯环与ligand1分子的喹啉环相互平行。经计算,平面间的距离为 (0.446±0.049) nm,符合产生π-π作用的条件。通过VMD跟踪观察,也发现FAM在模拟过程中一直保持这一结构。结合表3的能量分解分析,可以进一步确定,这种结合模式非常稳定,在分子识别和维持抑制活性方面作出了重要的贡献。
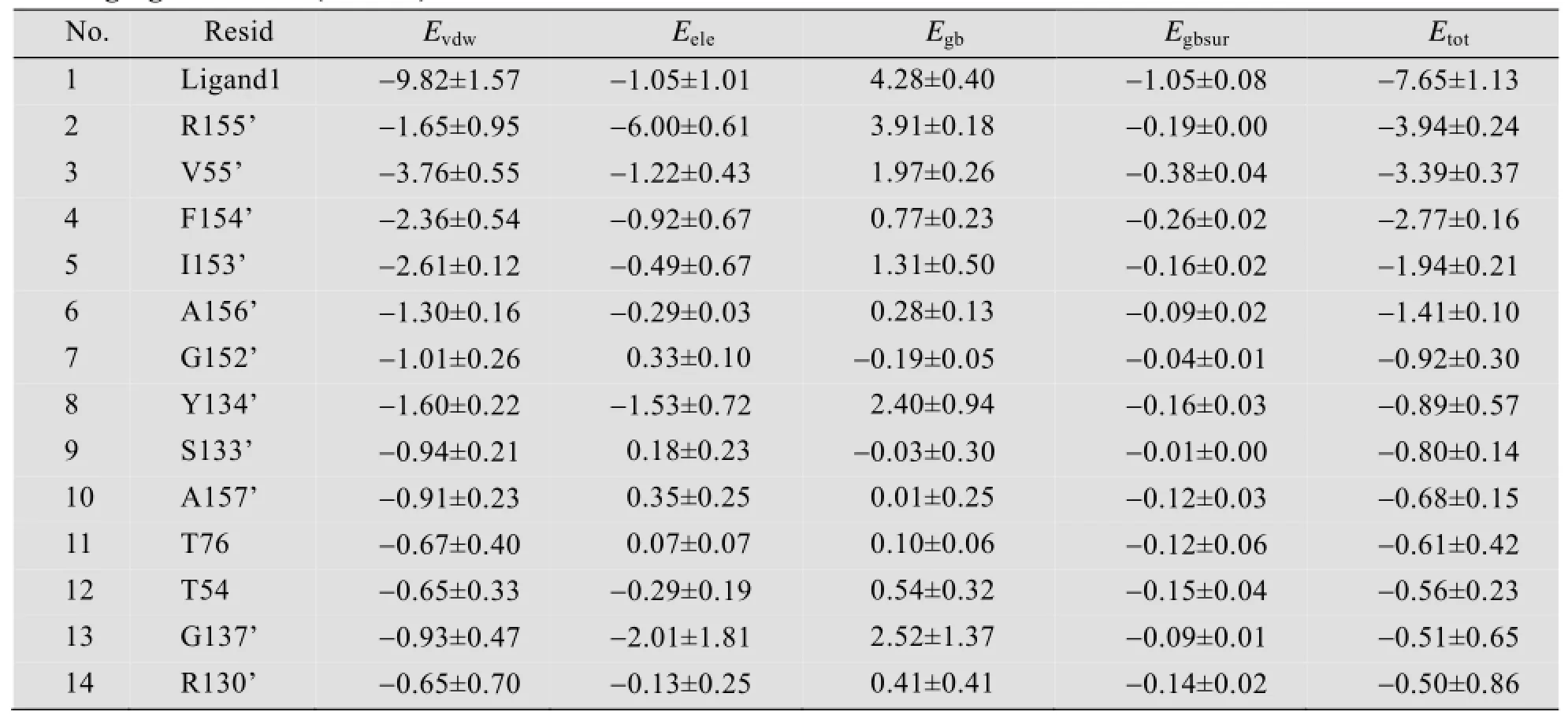
表4 HCV NS3/4A蛋白酶与底物FAM结合密切相关残基的能量分解结果 (kJ/mol)Table 4 The energy decomposition of the key residues in HCV NS3/4A protease significantly concerned with binding ligand FAM. (kJ/mol)
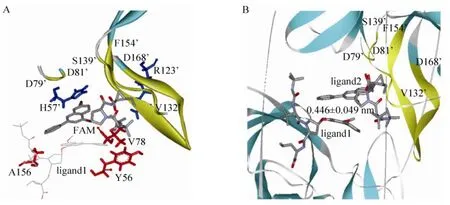
图6 HCV NS3/4A PR与底物FAM的结合模式图Fig. 6 The map of the binding mode between HCV NS3/4A protease and FAM.
2.4耐药性突变
有报道[32-35]显示,HCV NS3蛋白R155K、D168E/V和V170T突变株对包括Faldaprevir等多种HCV NS3/4A PR可逆抑制剂产生耐药。为了进一步探究这些突变残基对FAM分子识别的影响,对复合物晶体结构相关氨基酸进行了定点突变,并对突变后与FAM形成复合物的结合自由能量变进行了分析。图7给出了HCV NS3/4A PR氨基酸定点突变后关键残基对结合自由能贡献的变化。图中蓝色表示野生型 (Wild type,WT) HCV NS3/4A PR关键残基的结合自由能;红色表示R155K突变株NS3/4A PR关键残基的结合自由能;淡蓝色表示D168E突变株NS3/4A PR关键残基的结合自由能,而紫色和绿色则分别表示D168V和V170T突变株NS3/4A PR关键残基的结合自由能。
经计算,形成FAM-R155K突变株复合物的结合自由能变为-11.55 kJ/mol,FAM-D168E突变株复合物为-17.48 kJ/mol,而形成FAM-D168V突变株复合物与FAM-V170T突变株复合物的结合自由能变则分别为-10.91 kJ/mol和-12.14 kJ/mol。与野生型FAM-NS3/4A PR复合物的-31.73 kJ/mol相比不难看出,R155、D168和V170的突变对FAM的结合能力造成了不同程度的下降。说明FAM对突变型的HCV NS3/4A PR结合能力较弱。另外,从图7可以看出突变株的V55、I153和F154能量下降最明显。这可能是由于R-K、D-E、D-V以及V-T突变后,改变了口袋内部的疏水性造成的;此外,侧链体积增加,使得口袋内部的原子堆积,从而使得口袋发生了形变,降低了FAM的结合自由能。虽然突变株的A156、A157的结合自由能较野生型高,但由于突变后影响了FAM识别的大部分关键残基,故复合物体系总体自由能显著降低。再次证明了V55、I153和F154 对FAM的识别十分重要,与前文分析结果一致。
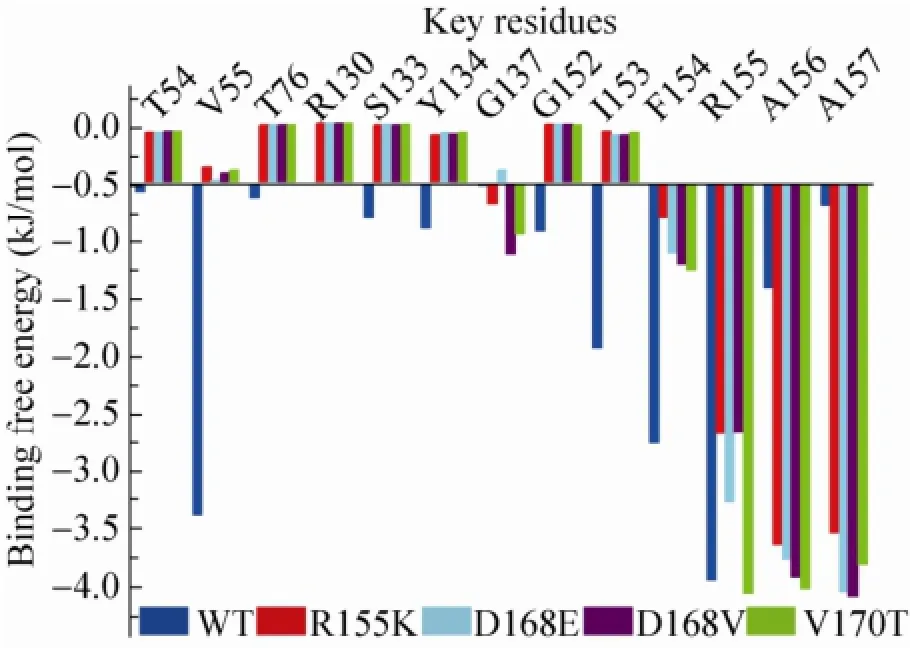
图7 HCV NS3/4A PR氨基酸定点突变后关键残基对结合自由能贡献的变化Fig. 7 The change of the decomposition of key HCV NS3/4A PR residues after the site-directed amino acid mutation.
2.5构象变化
2.5.1自由能曲面分析
图8给出了26.85 ℃下体系自由能曲面图。图中的颜色越深,相应区域的自由能越低。由图8可知,体系在模拟过程中存在4个相对独立的低自由能区域,即左侧的M1、右上部的M2、中部的M3以及右下部的M4。为了进一步分析图中出现低自由能区的原因,我们进行了聚类分析。这4个区域分别与图9中的4类能量最低构象相对应。
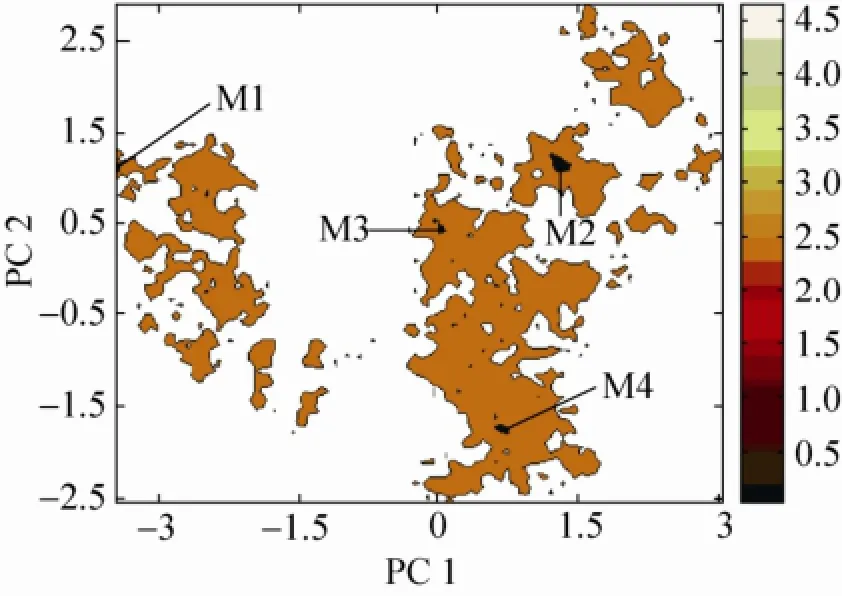
图8 体系的自由能曲面图Fig. 8 The free energy landscape map of the system.
2.5.2聚类分析
图9给出了HCV NS3/4A蛋白酶与FAM所构成的复合物聚类分析的结果。RMSD域值为0.31 nm。从图9中可以看出经过MD模拟所得的20 400个体系构象共4类,1.0 ns以前为第1类;2.76-4.41 ns、4.62-5.92 ns、6.03-6.04 ns、6.16-6.19 ns之间为第2类;4.42-4.61 ns、1.62-1.76 ns、5.93-6.15以及6.20-20.4 ns之间为第3类;剩余构象合为第4类,它们所占的概率分别是5.34%、15.63%、70.67%和8.36%。聚类分析结果表明,在MD模拟的20.4 ns时间内,HCV NS3/4A蛋白酶和配体FAM形成的复合物存在4个优势构象;各类构象之间有一定的结构跃迁,这也部分地解释了RMSD中出现的涨落。
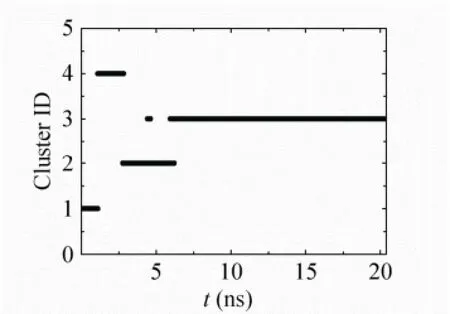
图9 基于HCV NS3/4A蛋白酶RMSD的聚类分析Fig. 9 Cluster analysis based on RMSD of the HCV NS3/4A protease.
图10给出了复合物聚类分析得到的各类代表性构象。图中结合于A链的FAM用stick模式表示;另一分子FAM和蛋白酶催化中心(H57、D81和S139) 用紫红色stick表示;蓝色片段表示能量分解中有较大贡献的F154-A157 和R130-Y134。从图10可以看到,在4类代表性构象中,口袋区的结合模式变化不大,仅是ligand2 (FAM) 分子的苯环与ligand1分子的喹啉环空间相对位置略有变化,这点在图6也有提到。结合模式方面,FAM的吡咯环主体通过C22和N23位取代侧链,稳定附着于F154-A157段,为其喹啉环侧链和乙烯基侧链顺利嵌入催化中心提供了保证。由于此3处催化中心位于蛋白酶内部,在模拟过程中,没有发生明显的相对位移,使得FAM更有效地占据催化中心,发挥抑制活性。
3 结论
对HCV NS3/4A蛋白酶与底物FAM的复合物进行20.4 ns的显含水分子动力学模拟。基于RMSF计算值找出了分子的柔性区域,并与实验B因子值进行了相关性分析,良好的相关性验证了模拟方法的可靠性。FAM的取代尿素侧链上的NH与蛋白酶A157,以及乙烯基侧链的O与S139形成较为稳定的氢键。结合自由能计算值与类似化合物的实验值吻合较好,其中范德华相互作用为复合物形成的主要驱动力。通过能量分解分析发现,FAM的双分子结合模式为发挥药效关键形式,V55在形成复合物的关键残基,对现有实验数据做了有益补充。随后的耐药性突变实验也证明了V55、I153和F154对FAM识别重要作用。最后,对复合物体系的构象变化做了简要分析,得到了模拟过程中的4类代表性稳定构象。模拟结果为以HCV NS3/4A蛋白酶为靶点的药物分子设计和药物分子的作用机理研究提供了一定的理论指导。
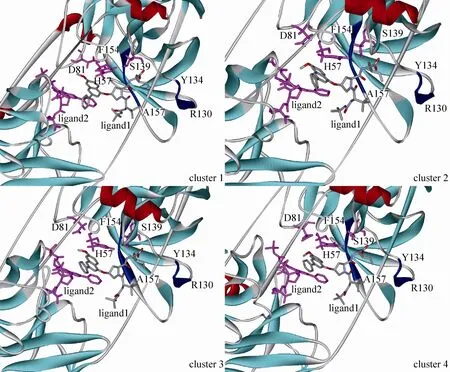
图10 聚类分析的代表构象Fig. 10 The representative conformation of cluster analysis.
REFERENCES
[1] Guan Y, Sun HY, Li YY, et al. The competitive binding between inhibitors and substrates of HCV NS3/4A protease: a general mechanism of drug resistance. Antiviral Res, 2014, 103 (3): 60–70.
[2] Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect, 2011, 17(2): 107–115.
[3] Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis, 2005, 5(9): 558–567.
[4] Chen SL, Morgan TR. The natural history of hepatitis C virus (HCV) infection. Int J Med Sci, 2006, 3(2): 47–52.
[5] Poordad F, Dieterich D. Treating hepatitis C: current standard of care and emerging direct-acting antiviral agents. J Viral Hepat, 2012, 19(7): 449–464.
[6] Zeuzem S, Poordad F. Pegylated-interferon plus ribavirin therapy in the treatment of CHC: individualization of treatment duration according to on-treatment virologic response. Curr Med Res Opin, 2010, 26(7): 1733–1743.
[7] Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology, 2014, 146(5): 1176–1192.
[8] Kim DW, Gwack Y, Han JH, et al. C-terminaldomain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem Biophys Res Commun, 1995, 215(1): 160–166.
[9] Baril M, Racine ME, Penin F, et al. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol, 2009, 83(3): 1299–1311.
[10] Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA, 2005, 102(8): 2992–2997.
[11] Foy E, Li K, Wang CF, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science, 2003, 300(5622): 1145–1148.
[12] LaPlante SR, nar H, Lemke CT, et al. Ligand bioactive conformation plays a critical role in the design of drugs that target the hepatitis C virus NS3 protease. J Med Chem, 2014, 57(5): 1777–1789.
[13] Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules. J Comput Chem, 2000, 21(12): 1049–1074.
[14] Wang JM, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem, 2004, 25(9): 1157–1174.
[15] Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys, 1983, 79(2): 926–935.
[16] Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys, 1977, 23(3): 327–341.
[17] Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res, 2000, 33(12): 889–897.
[18] Wang W, Donini O, Reyes CM, et al. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophys Biomol Struct, 2001, 30(1): 211–243.
[19] Simonson T. Macromolecular electrostatics: continuum models and their growing pains. Curr Opin Struct Biol, 2001, 11(2): 243–252.
[20] Bashford D, Case DA. Generalized born models of macromolecular solvation effects. Annu Rev Phys Chem, 2000, 51 (1): 129–152.
[21] Still WC, Tempczyk A, Hawley RC, et al. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc, 1990, 112(16): 6127–6129.
[22] Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem, 1999, 20(2): 217–230.
[23] Maisuradze GG, Liwo A, Scheraga HA. Relation between free energy landscapes of proteins and dynamics. J Chem Theory Comput, 2010, 6(2): 583–595.
[24] Chen LY. Exploring the free-energy landscapes of biological systems with steered molecular dynamics. Phys Chem Chem Phys, 2011, 13(12): 6176–6183.
[25] Nguyen PH, Derrenumaux P. Configurational entropy: an improvement of the quasiharmonic approximation using configurational temperature. Phys Chem Chem Phys, 2012, 14(2): 877–886.
[26] Hegger R, Altis A, Ngeuyen PH, et al. How complex is the dynamics of peptide folding? Phys Rev Lett, 2007, 98(2): 028102.
[27] Barlow PN, Steinkasserer A, Norman DG, et al. Solution structure of a pair of complement modules by nuclear magnetic resonance. J Mol Biol, 1993, 232(1): 268–284.
[28] Feig M, Karanicolas J, Brooks III CL, et al. MMTSB tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graphics Modell, 2004, 22(5): 377–395.
[29] Daura X, Gademann K, Jaun B, et al. Peptide folding: when simulation meets experiment. Angew Chem Int Ed, 1999, 38(1/2): 236–240.
[30] Hu JP, Gong XQ, Su JG, et al. Study on the molecular mechanism of inhibiting HIV-1 integrase by EBR28 peptide via molecular modeling approach. Biophys Chem, 2008, 132(2/3): 69–80.
[31] Welsch C, Schweizer S, Shimakami T, et al. Ketoamide resistance and hepatitis C virus fitness in Val55 variants of the NS3 serine protease. Antimicrob Agents Chemother, 2012, 56(4): 1907–1915.
[32] Berger KL, Lisetté L, Triki I, et al. Viral resistance in hepatitis C virus genotype 1-infected patients receiving the NS3 protease inhibitor Faldaprevir (BI 201335) in a phase 1b multiple-rising-dose study. Antimicrob Agents Chemother, 2013, 57(10): 4928–4936.
[33] Berger KL, Triki I, Cartier M, et al. Baseline hepatitis C virus (HCV) NS3 polymorphisms and their impact on treatment response in clinical studies of the HCV NS3 protease inhibitor faldaprevir. Antimicrob Agents Chemother, 2014, 58(2): 698–705.
[34] zeuzem S, buggisch P, agarwal K, et al. The protease inhibitor, GS-9256, and non-nucleoside polymerase inhibitor tegobuvir alone, with ribavirin, or pegylated interferon plus ribavirin in hepatitis C. Hepatology, 2012, 55(3): 749–758.
[35] Soumana DI, Ali A, Schiffer CA. Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS Chem Biol, 2014, 9(11): 2485–2490.
(本文责编 郝丽芳)
Molecular recognition mechanism and motion of HCV NS3/4A protease with Faldaprevir analogue
Li Liang1, Jianping Hu1,2, Wenyi Du1, Ke Zuo1, Wei Liu1, and Xiaojun Gou1
1 Key Laboratory of Medicinal and Edible Plants Resources Development of Sichuan Education Department, Sichuan Industrial Institute of Antibiotics, Chengdu University, Chengdu 610106, Sichuan, China
2 College of Chemistry, Leshan Normal University, Leshan 614004, Sichuan, China
Abstract:Faldaprevir analogue molecule (FAM) has been reported to effectively inhibit the catalytic activity of HCV NS3/4A protease, making it a potential lead compound against HCV. A series of HCV NS3/4A protease crystal structures were analyzed by bioinformatics methods, and the FAM-HCV NS3/4A protease crystal structure was chosen for this study. A 20.4 ns molecular dynamics simulation of the complex consists of HCV NS3/4A protease and FAM was conducted. The key amino acid residues for interaction and the binding driving force for the molecular recognition between the protease and FAM were identified from the hydrogen bonds and binding free energy analyses. With the driving force of hydrogen bonds and van der Waals, FAM specifically bind to the active pocket of HCV NS3/4A protease, including V130−S137, F152−D166, D77−D79 and V55, which agreed with the experimental data. The effect of R155K, D168E/V and V170T site-directed mutagenesis on FAM molecular recognition was analyzed for their effect on drug resistance, which provided the possible molecular explanation of FAM resistance. Finally, the system conformational change was explored by using free energy landscape and conformational cluster. The result showed four kinds of dominant conformation, which provides theoretical basis for subsequent design of Faldaprevir analogue inhibitors based on the structure of HCV NS3/4A protease.
Keywords:HCV NS3/4A protease, Faldaprevir analogue molecule, molecular dynamics simulation, binding free energy, conformational change
Corresponding authors: Jianping Hu. Tel: +86-833-2152297; E-mail: hujianping@emails.bjut.edu.cn
