基于密度泛函理论的尼克酸分子振动光谱分析
2016-06-30陶亚萍韩礼刚韩运侠刘照军
陶亚萍,韩礼刚,韩运侠,刘照军
(洛阳师范学院物理与电子信息学院,洛阳 471022)
基于密度泛函理论的尼克酸分子振动光谱分析
陶亚萍,韩礼刚,韩运侠,刘照军*
(洛阳师范学院物理与电子信息学院,洛阳471022)
摘要:实验测量了尼克酸分子的拉曼和红外光谱,用B3LYP混合泛函和cc-PVDZ基组计算了尼克酸分子的平衡构型、振动频率、拉曼和红外强度。采用GAR2PED程序对尼克酸分子进行了简正振动分析,依据所得势能分布对尼克酸分子的振动频率进行了理论归属,弥补了以往文献缺少对其振动模式贡献进行定量研究的不足,提供了更多的振动光谱信息。
关键词:尼克酸;密度泛函理论;振动光谱;简正振动分析;频率归属
1引言
尼克酸(Niacin,C6H5NO2)为白色或淡黄色结晶性粉末,属于水溶性B族维生素,与烟酰胺一起合称为维生素PP。最初它是通过氧化尼古丁得到的,因此也称为尼古丁酸。从结构上看,它是由吡啶在3号位置连接一个羧基组成,亦称为吡啶-3-甲酸。化学上一般把具有生物活性的全部吡啶-3-甲酸及其衍生物统称为烟酸。尼克酸在自然界中分布很广,尤其在动物肝脏、酵母、蛋黄、谷物、花生、鱼类和绿色蔬菜中含量较高。它被广泛用于食品、化学和医药等行业中。在食品工业中,可用做去味剂、防腐剂、保色剂和保鲜剂等;在饲料加工行业中,可提高鸡鸭的产蛋率、奶牛产奶量及鱼、牛、羊等禽畜的抗病能力和肉的质量;在日用化学工业中,它可用于合成头发生长促进剂、染发助剂、聚合物稳定剂、感光材料的抗氧化剂与抗灰雾剂、电镀光亮剂及PVC塑料的热稳定剂等;在医药行业中,它有促进细胞新陈代谢、扩张血管、缓解末稍血管痉挛的效果并且有保护皮肤中的胶原纤维的功效,可用于防治糙皮病和类似的维生素缺乏症、动脉硬化等症,它作为医药中间体可用于合成治疗中枢性呼吸及循环系统衰竭症、偏头痛、高血酯症、冠心病等疾病的烟酸铬、烟酰胺、尼克刹米、烟酸肌醇脂、癸烟酯等数十种药物[1-3]。因此对尼克酸的研究具有重要的现实意义。
目前有关尼克酸的研究主要集中在其含量的色谱法测定、晶体结构[4-5]、二聚体结构与性质的分析方面[6],同时对尼克酸的表面增强拉曼光谱也有较多的研究[7-10]。振动光谱是研究分子结构的重要手段,而密度泛函理论可以从理论上对分子的振动光谱进行预测,因此密度泛函理论在分子光谱的研究领域中应用越来越普遍[11-12]。在尼克酸的振动光谱分析方面,一直备受关注,已有一些相关的研究,比如:2001年,O.Sala测量了尼克酸水溶液1800~200 cm-1波段的拉曼光谱,并对此波段的振动模式进行了初步的指认[13];2003年,P.Koczoń给出了尼克酸固体粉末3500~3000和1800~1300 cm-1波段的红外、拉曼光谱的实验频率及理论归属[14];2004年,莫卫民等人对尼克酸的红外光谱实验结果进行了初步分析[15];2006年,Li-Ran Wang测量了尼克酸溶液1700~400 cm-1波段的拉曼光谱,并指出当溶液呈中性或弱碱性时,尼克酸以离子形式存在于溶液中[16];2009年,韩礼刚也曾结合Gauss View[17]软件显示的分子振动的动画对尼克酸的拉曼光谱的振动频率进行了初步归属[18];2011年,M.Kumar和R.A.Yadav对尼克酸及其N-氧化物的振动光谱进行了对比分析,也结合Gauss View软件给出了经验的归属[19]。虽然相关研究较多,但未见到结合密度泛函理论与简正振动分析程序对尼克酸的拉曼、红外光谱的全部波段进行全面归属的文献,前人的讨论均未能定量的给出每种基团特征振动模式的贡献百分比。基于此,在实验测定尼克酸分子的红外和拉曼光谱的基础上,结合密度泛函理论和简正振动模式分解的分析方法,来获得各基团特征振动模式的势能分布,从而对尼克酸分子的振动光谱进行了比较全面的归属。
2实验
实验中用于测试的尼克酸样品购于北京化工厂,纯度大于98%,未经进一步提纯。红外光谱采用的是KBr压片制作,使用BIO-RAD 60 V 型傅立叶变换红外光谱仪测量,光谱分辨率为1 cm-1。拉曼光谱使用LabRam HR800型共焦显微拉曼光谱仪测量,激光光源为氩离子激光器的514.5 nm,到达样品表面的功率为3 mW,50倍物镜,1800 gr/mm的高分辨率闪耀衍射光栅,数据采集时间为30秒。
3计算方法
尼克酸是由吡啶在3号位被羧酸基团取代而成,虽然只有1个取代基团,但由于羧酸基团相对于吡啶环、羟基基团相对于羰基基团朝向的不同,尼克酸会出现4种构型。为了找到尼克酸分子的最稳定构型,采用Gaussian 09[20]程序包、密度泛函理论方法(DFT)和B3LYP/6-311G(d,p)基组,计算了4种构型的能量和频率,发现除了(d)构型有虚频外,其它三种构型均无虚频。图1是计算所得尼克酸分子4种构型的示意图。每种构型对应的能量分别为:(a)-436.865910 a.u.;(b)-436.865554 a.u.;(c)-436.853413 a.u.;(d)-436.855636 a.u.,通过比较,可以看到构型(a)能量最低、最稳定,李会学等人[6]通过过渡态的分析也确定了构型(a)最稳定,同时M.P.Gupta[4]、W.B.Wright[5]等人报道的晶体结构也得出了同样的结论,故此本文以下分析和讨论是基于构型(a)展开的。
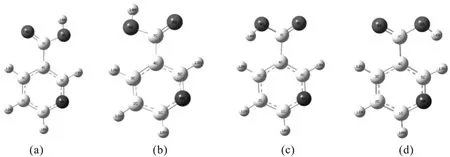
Fig.1 The optimized equilibrium geometry of Niacin
为了对简正振动频率做出正确的归属,采用更大的基组B3LYP/cc-PVDZ对构型(a)进行了进一步的结构优化和频率计算。尼克酸分子共有14个原子,42个笛卡尔坐标和36个简正振动频率。根据分子的对称性和P.Pulay提出的方法[21-22],构造出一套共36个独立、完备的局域对称坐标,如表1所示。最后由GAR2PED 软件[23]读取Gaussian程序的输出文件,得到每个简正振动模式在各对称坐标上的势能分布百分比,判断出每种基团特征振动模式的贡献,从而完成各简正频率的归属指认。
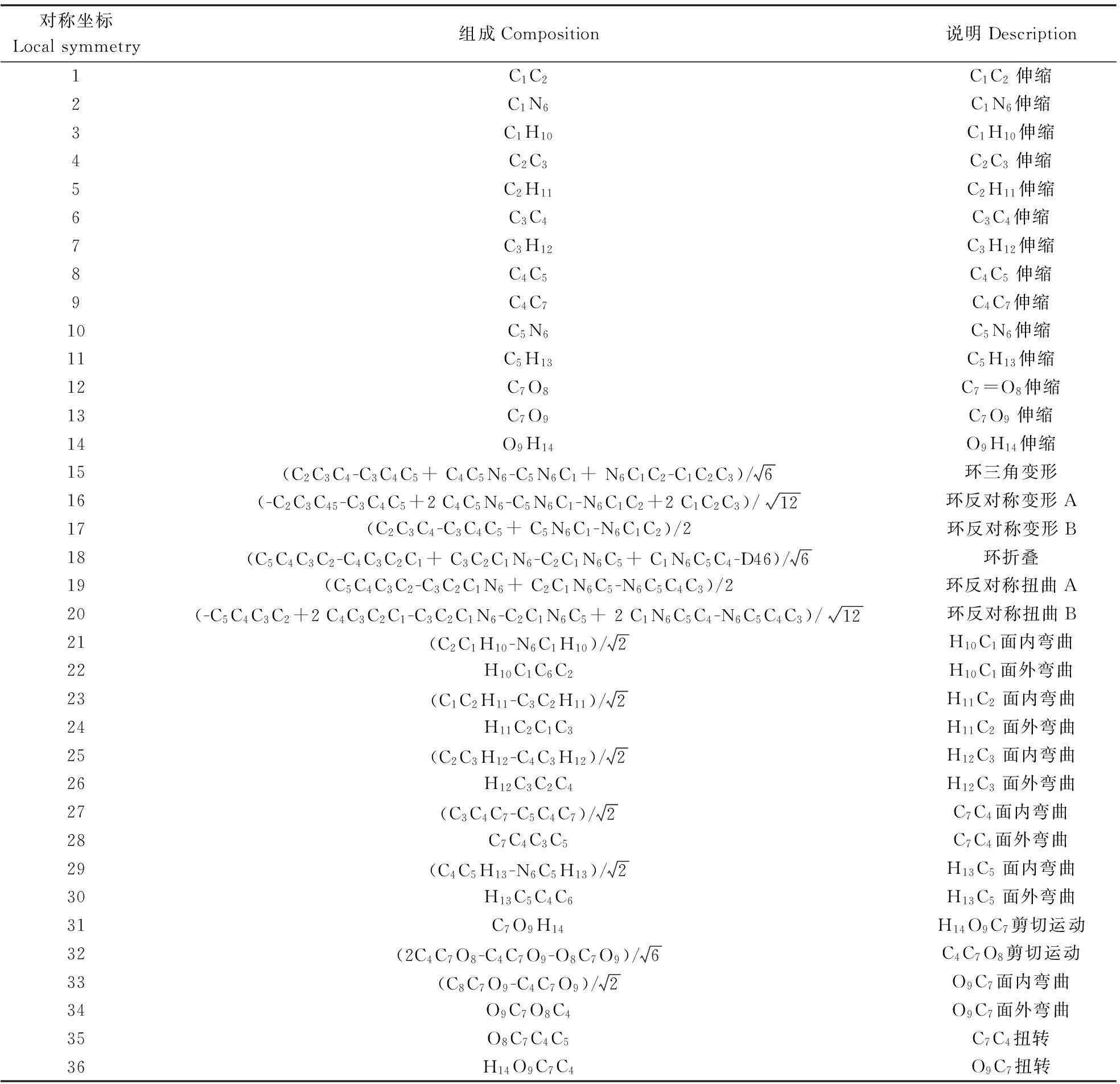
Tab.1 Local symmetry coordinates of Niacin
4结果与讨论
4.1尼克酸分子的平衡构型
据我们所知,尼克酸分子的结构在气相中尚未确定,但固体状态的晶体结构已经由M.P.Gupta等人[4]进行了报道,理论和实验结构参数的对比如表2所示。尼克酸是一个含氮原子的六元杂环化合物,是吡啶衍生物的一种。由于存在氮原子,使得分子中的六元环状结构略显不规则,C1-N6、C5-N6键长较C1-C2、 C2-C3、C3-C4、C4-C5略短;键角C4-C5-N6、N6-C1-C2略大于C1-C2-C3、C2-C3-C4、C3-C4-C5、C5-N6-C1。换言之,吡啶环上围绕着氮原子的键长和键角与苯环相比,均有较大变化。这一方面是因为氮原子自身的影响,另一方面是因为羟基与氮原子形成O-H…N分子间氢键的缘故。
忽略羧基,吡啶环上C-C、C-N键长的理论值与实验值的均方根偏差(RMSD)是0.019 Å,键角的偏差是0.72°。考虑羧基,尼克酸分子键长的均方根偏差是0.028 Å,键角的偏差是1.82°。偏差最大的两个角是C4-C7-O8和C4-C7-O9,这是因为由于分子间的相互作用,在固态中尼克酸的羧基会形成O-H…O氢键,会对羧基的键长和键角产生较大的影响,这也是造成均方根偏差明显增大的主要原因。同时C4-C7单键的键长实验值比标准的C-C单键键长(1.54 Å)明显缩短,C7-O9键长的实验值明显比理论值小,亦可证明O-H…O氢键的存在。正是由于固体状态中分子间会形成O-H…O和O-H…N两种氢键,而理论计算的是单分子的气态平衡构型,未考虑到氢键的影响,所以理论计算的结果与实验数据会稍微有一些偏差。从表2的计算结果可以看到,所有的二面角都是0.0° 或180.0°,即羧酸基团和吡啶环是共面的,表明尼克酸分子是平面结构。

Tab.2 Geometry parameters of Niacin
4.2振动频率和归属
为了对光谱图进行直观的比较,我们根据文献[24-25]把Gaussian09计算所得的拉曼活性Si转换为谱峰的强度Ii,其理论和实验光谱的比较如图2和3所示。计算结果表明尼克酸分子的36个简正振动由25个面内振动和11个面外振动组成,具有Cs对称性,所有振动模既有拉曼活性又有红外活性。为了使密度泛函理论获得的理论值与实验值更加吻合,J.P.Merrick 等人[26]在充分考虑电子结构、非谐性等影响的基础上,通过1066个理论频率与相应实验频率的对比计算和分析,得出了B3LYP/cc-PVDZ 算法的频率校正因子为0.9717。于是笔者把尼克酸分子的理论计算频率与该校正因子相乘,经过校正后,计算值与实验观测值较好的吻合,其中存在的少许差别可能来自于理论模拟的是单分子理想化模型,而实验测量的样品存在分子间相互作用,如表3所示。同时表3显示了由简正振动分析软件得到的各种基团特征振动模式的贡献百分比(势能占比大于5%的振动)。
通常自由的O-H键伸缩振动应该出现在3500 cm-1以上[27],但在尼克酸中O-H键与C=O双键是相连的,在羰基和分子间形成的O-H…O氢键的双重影响下,O-H键伸缩的振动频率会减小。 因此在本研究中,我们将位于3416 cm-1的红外峰归属为O-H伸缩振动,是比较合理的。尼克酸分子属于单取代杂环芳香烃分子,因此它应该有4个C-H伸缩振动,且它们均属于典型的含氮杂环芳香烃的C-H伸缩振动,应该出现在3100~3010 cm-1区域[27],它们的振动相对独立,与其它键伸缩或弯曲振动之间耦合较小,而本研究中位于3088、 3073、3042 cm-1的拉曼峰和位于3097、3071、3041 cm-1的红外峰被指认为C-H伸缩振动,与文献报道的范围基本一致;位于1708 cm-1的红外强峰和1694 cm-1的拉曼强峰,从势能分布的数值可以看到C=O伸缩振动的比例高达92%,因此被归属为C=O伸缩振动,这与文献[28]报道的羧基的特征基团C=O伸缩振动通常位于1715~1680 cm-1是一致的。事实上,M.Kumar和R.A.Yadav[19]根据Gauss View软件的动画将位于3414 cm-1的红外强峰归属为尼克酸分子的O-H伸缩振动,将位于3089、3073、3041 cm-1的拉曼峰与位于3099、3075、3040 cm-1的红外峰归属为C-H伸缩振动,将位于1711 cm-1的红外强峰和1695 cm-1的拉曼强峰归属为C=O伸缩振动,这与我们的归属都是一致的,可见对这种相对独立、耦合较小的振动,归属并不存在争议,差别在于我们是通过特征基团贡献百分比的数值进行了定量化的归属。
分子的振动是分子中所有原子共同参与的简正振动,很多情况下表现为多个振动耦合的形式。对于尼克酸分子的振动,在频率较低的部分(<1700 cm-1),这种耦合现象就很普遍。M.Kumar和R.A.Yadav[19]指出对于这种强烈耦合振动的归属是非常困难的,因此他们只给出了部分经验归属,未展现耦合振动部分。而本文依据简正振动分析方法对尼克酸分子的振动频率能够进行全面的归属,展现了耦合振动过程中各基团振动的贡献百分比。如:从表3很容易看出来位于1417、1309、1187、1085 cm-1处的拉曼峰和位于1416、1297、1186、1088 cm-1处的红外峰归属于C-H面内弯曲振动,其中含有C-C伸缩振动的成分;位于1002、975、954、812 cm-1处的拉曼峰和954、813 cm-1处的红外峰归属于C-H面外弯曲振动;同时位于1600、1589 cm-1处的拉曼峰和1599、1583 cm-1处的红外峰归属于C-C伸缩振动,这与以往文献[29]报道环的C-C伸缩振动一般应出现在1625~1430 cm-1范围内是一致的;位于1321、1242 cm-1处的拉曼峰和1322、1243 cm-1处的红外峰归属于C-N伸缩振动,亦耦合有C-C伸缩振动成分;位于1115 cm-1处的拉曼和红外峰归属于C-O伸缩振动;位于1031、751 cm-1处的拉曼峰和1039 cm-1处的红外峰归属于环面内变形,695、387 cm-1处的拉曼峰和695 cm-1处的红外峰归属于环面外变形振动。与P.Koczoń[16]的理论归属相比,差别主要在苯环上的C-C和C-N伸缩振动的归属上。P.Koczoń[14]把环上各键的伸缩振动统一称为环伸缩振动,没有具体给出环上哪个键的伸缩振动所占比例更多一些,而我们给出的势能分布结果很清晰的给出了各个振动模式所占的比例。
5结论
用量子化学方法计算了尼克酸分子的4种可能构型和相应的能量,通过能量的对比、文献调查和实验的佐证确定了尼克酸分子的最稳定构型,计算了最稳定构型的简正振动频率和相应的红外与拉曼强度,并对各简正振动频率进行了全面归属。尼克酸分子有两个氢键受体(-N、C=O)和一个供体(-OH),在固体状态下存在O-H…O和O-H…N两种氢键,会对其结构和光谱产生一定的影响。M.Kumar和R.A.Yadav[19]曾经指出,因为环上各键振动之间存在耦合,与取代基的振动也存在耦合,因此对尼克酸的吡啶环振动的归属以及与环相连的C-H键振动的归属都是非常困难的。而本文依据的简正振动分析方法,通过比较势能分布百分比的方式,可以容易地了解由哪种特征振动模式所主导,这种定量方法比以往的经验归属更准确。P.Koczoń[14]虽然也采用了简正振动分析方法,但它只给出了部分波段的归属,而且没有具体给出吡啶环上各振动的贡献比例。本文利用简正振动分析方法给出了尼克酸分子全部谱带的势能分布,并对其进行了全面的归属。尼克酸广泛用于食品、化学和医药等行业中,对该物质分子结构的分析和振动频率的全面归属,为与其相关的药学、生物学等方面的深入研究提供了重要的参考价值。
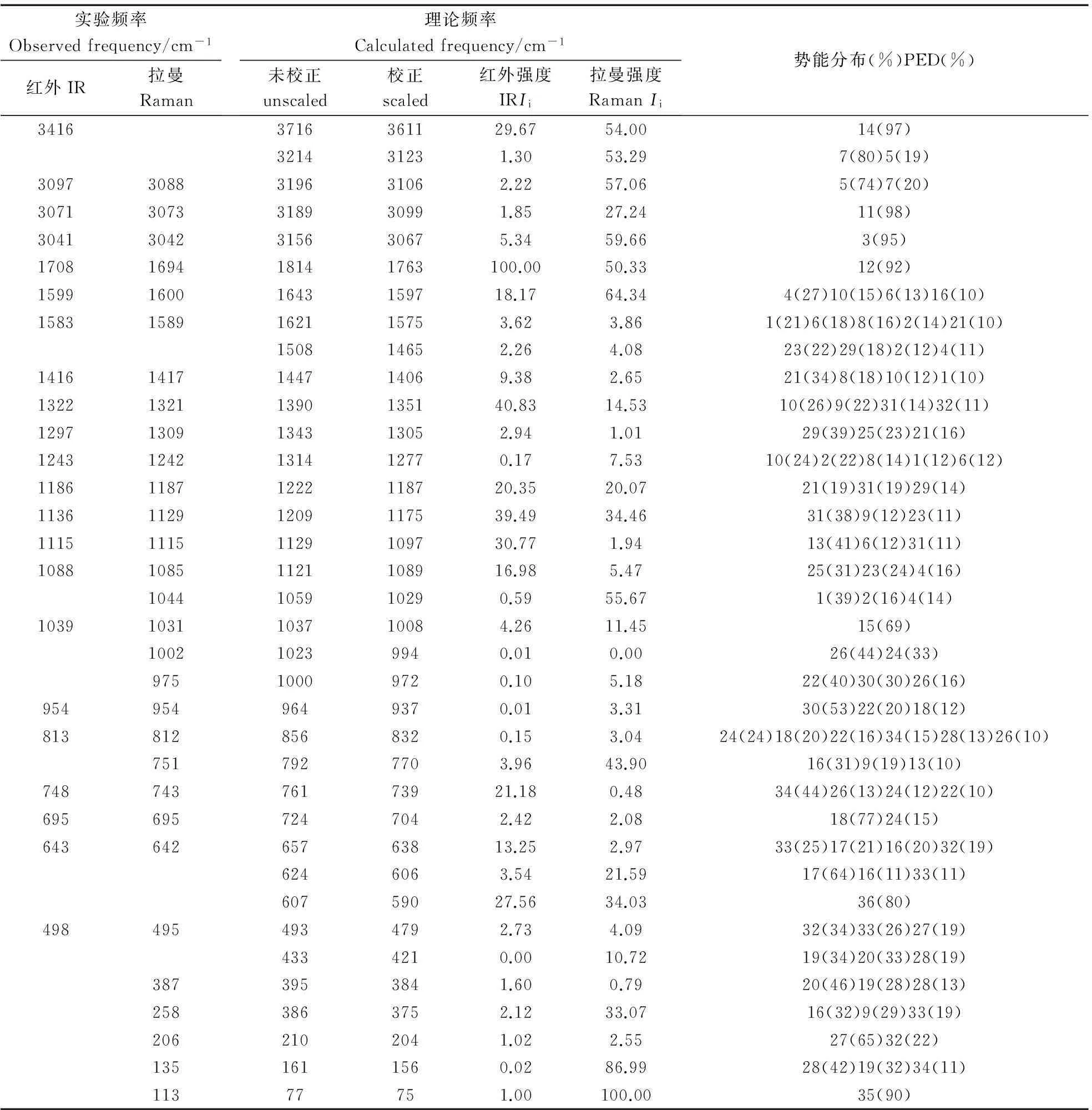
Tab.3 Calculated and observed vibrational frequencies with PED of Niacin
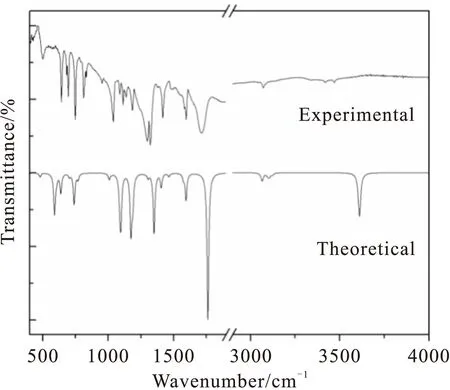
Fig.2 Infrared spectra of Niacin

Fig.3 Raman spectra of Niacin
参考文献
[1]赵东江,马松艳.烟酸的合成方法与开发前景[J].江苏化工,2005,33(1):53-57.(Zhao Dongxiang,Ma Songyan.Synthesis Methods and Development Prospects of Nicotinic Acid [J].Jiangsu Chem Ind,2005,33(1):53-57.)
[2]路百程.烟酸的合成与应用[J].辽宁化工,1994,3:9-12.(Lu Baicheng,Synthesis and Application of Nicotinic Acid [J].Liaoning Chem Ind,1994,3:9-12.)
[3]李艳云,尹振晏,宫彩红.烟酸的研究进展[J].北京石油化工学院学报,2006,14(1):59-64.(Li Yanyun,Yin Zhenyan,Gong Caihong.Development of Nicotinic Acid [J].J Beijing Ins.Petrochem Tech,2006,14(1):59-64.)
[4]Gupta M P,Kumar P.Nicotinic Acid:a Redetermination [J].Cryst Struct Commun,1975,4:365.
[5]Wright W B,King G S.The crystal structure of nicotinic acid [J].Acta Crystal,1953,6(4):305-317.
[6]李会学,王晓峰,董小宁,等.烟酸二聚体的结构与性质[J].物理化学学报,2009,25(1):161-168.(Li Huixue,Wang Xiaofeng,Dong Xiaoning.etal.Structures and Properties of Nicotinic Acid Dimer [J].Acta Phys-Chim Sin,2009,25(1):161-168.)
[7]Park S M,Kim K.Adsorption of picolinic and niacins on a silver sol surface investigated by Raman spectroscopy [J].J Mol Stru,1995,344:195-203.
[8]Baia M,Astilean S.Raman and Sers Investigations of Pharmaceuticals [M].Traian Iliescu Published by Springer,2008:137-142.
[9]Zhang L S,Fang Y,Zhang P.Experimental and DFT theoretical studies of SERS effect on gold nanowires array [J].Chem Phy Lett,2008,451(1):102-105.
[10]Wen R,Fang Y.Adsorption of pyridine carboxylic acids on silver surface investigated by potential-dependentSERS [J].Vib Spec,2005,39(1):106-113.
[11]陶亚萍,吴义芳,韩礼刚,等.柠檬酸分子振动光谱研究[J].光散射学报,2015,27(3):256-262.(Tao Yaping,Wu Yifang,Han Ligang,etal.Investigation on the vibrational spectra of citric acid [J].J Light Scatter,2015,27(3):256-262)
[12]陶亚萍,韩礼刚,梁会琴,等.2-巯基-5-甲基-1,3,4-噻二唑分子的拉曼、红外光谱和简正振动分析[J].光散射学报,2012,24(4):381-387.(Tao Yaping,Han Ligang,Liang Huiqin,etal.Raman,FT-IR Spectra and Normal Mode Analysis of 2-Mercapto-5-methyl-1,3,4-thiadiazole [J].J Light Scatter,2012,24(4):381-387.)
[13]Sala O,Goncalves N S,Noda L K.Vibrational analysis of Nicotinic species based on ab initio molecular orbital calculations [J].J Mol Stru,2001,566:411-416.
[14]Koczoń P,Dobrowolski J C,Lewandowski W,etal.Experimental and theoretical IR and Raman spectra of picolinic,nicotinic and isoniacins [J].J Mol Stru,2003,655(1):89-95.
[15]莫卫民,薛建,胡宝祥,等.红外光谱在烟酸系列化合物制备过程中的应用[J].光谱实验室,2004,21(4):677-679.(Mo Weimin,Xue Jian,Hu Baoxiang,etal.The Application of FT-IR in the Synthesis of Nicotinic Derivatives [J].J Spec Lab,2004,21(4):677-679.)
[16]Wang L R,Fang Y.Experimental(UV-Raman)and theoretical(DFT)study of pyridine carboxylic acid in aqueous solution [J].Chem Phy,2006,323(2):376-382.
[17]Dennington R,Keith T,Millam J.GaussView 5.0.9[CP].Semichem Inc,Shawnee Mission,KS,2009.
[18]韩礼刚.烟酸分子的简正振动与拉曼光谱[J].洛阳师范学院学报,2009,28(2):44-46.(Han Ligang.Normal Vibration and Raman Spectrum of Nicotinic Acid [J].J Luoyang Norm Univ,2009,28(2):44-46.)
[19]Kumar M,Yadav R A.Experimental IR and Raman spectra and quantum chemical studies of molecular structures,conformers and vibrational characteristics of nicotinic acid and its N-oxide [J].Spectrochim Acta Part A,2011,79(5):1316-1325.
[20]Frisch M J,Trucks G W,Schlegel H B,etal.Gaussian 09 D.01[CP].Gaussian Inc,Wallingford CT,2013.
[21]Pulay P,Fogarasi G,Pang F,etal.Systematic ab Initio Gradient Calculation of Molecular Geometries,Force Constants,and Dipole Moment Derivatives [J].J Amer Chem Soc,1979,101(10):2550-2560.
[22]Pulay P,Paizs B.Newtonian molecular dynamics in general curvilinear internal coordinates [J].Chem Phy Lett,2002,353(5):400-406.
[23]Martin J M,Alsenoy C V.Gar2ped [CP].University of Antwerp,1995.
[24]Krishnakumar V,Surumbarkuzhali N,Muthunatesan S.Scaled quantum chemical studies on the vibrational spectra of 4-bromo benzonitrile [J].Spectrochim Acta,Part A,2009,71(5):1810-1813.
[25]Chalmers J M,Griffiths P R.Handbook of Vibrational Spectroscopy [M].John Wiley & Sons Ltd,2002,71.
[26]Merrick J P,Moran D,Radom L.An evaluation of harmonic vibrational frequency scale factors [J].J Phy Chem A,2007,111(45):11683-11700.
[27]Socrates G.Infrared and Raman characteristic group frequencies,tables and charts [M].Wiley Chichester,2001:68-235.
[28]Monicka J C,James C.FT-Raman and FTIR spectra,DFT investigation of the structure and vibrational assignment of mefenacet [J].J Mol Stru,2015,1095:1-7.
[29]Tao Y,Han L,Han Y,etal.A combined experimental and theoretical analysis on molecular structure and vibrational spectra of 2,4-dihydroxybenzoic acid [J].Spectrochim Acta,Part A,2015,137:1078-1085.
Analysis of Vibrational Spectra of Niacin Based on Density Functional Theory
TAO Ya-ping,HAN Li-gang,HAN Yun-xia,LIU Zhao-jun*
(CollegeofPhysicsandElectronicInformation,Luoyangnormaluniversity,Luoyang,471022)
Abstract:The Raman and infrared spectra of niacin were measured experimentally.The molecular equilibrium geometries,vibrational frequencies,Raman and infrared intensity of niacin were calculated using the B3LYP level and cc-PVDZ basis set.Normal mode analysis was carried out using the program GAR2PED and the assignment of fundamental vibrations for niacin was obtained according to the potential energy distributions(PED).This study provides us with more quantitative vibrational spectral information which was not mentioned in previous literatures.
Key words:niacin;DFT;vibrational spectrum;normal mode analysis;frequencies assignments
收稿日期:2015-07-30; 修改稿日期:2015-12-21
基金项目:NSFC-河南人才培养联合基金(U1204109)河南省科技计划项目(142102210109)
作者简介:陶亚萍(1980-),女,内蒙古通辽人,讲师,主要从事分子光谱的理论和实验研究。E-mail:taoyaping2001@gmail.com 通讯作者:刘照军(1966-),男,教授,从事拉曼光谱实验研究。E-mail:zhaojunliu@gmail.com
文章编号:1004-5929(2016)02-0175-07
中图分类号:O657.37
文献标志码:A
doi:10.13883/j.issn1004-5929.201602014
