布洛芬分子手性转变裸反应机理及水分子的催化作用
——基于羰基和苯环作H迁移桥梁*
2016-06-05王佐成闫红彦杨晓翠
高 峰,王佐成,闫红彦,杨晓翠,佟 华
(1.白城师范学院物理学院,吉林 白城 137000;2.白城师范学院计算机科学学院,吉林 白城 137000)
布洛芬分子手性转变裸反应机理及水分子的催化作用
——基于羰基和苯环作H迁移桥梁*
高 峰1,王佐成1,闫红彦2,杨晓翠1,佟 华1
(1.白城师范学院物理学院,吉林 白城 137000;2.白城师范学院计算机科学学院,吉林 白城 137000)
采用基于密度泛函理论的B3LYP方法和微扰理论的MP2方法,研究了布洛芬分子手性转变裸反应和水助质子从手性碳向羰基迁移的机理。分子结构分析表明:水助质子从手性碳向羰基迁移过程的8元环过渡态bTS2·2H2O和10元环过渡态bTS2·3H2O对应的氢键键角都远大于6元环过渡态bTS2·1H2O;过渡态bTS2·2H2O的8元环结构基本共面,过渡态a_TS1·3H2O和bTS2·3H2O的10元环结构明显偏离平面。反应路径研究发现:标题反应有6条路径,分别是质子只以羰基氧、以甲基碳和羰基O及以羧基和苯环联合作桥,从手性C的一侧迁移到另一侧。势能面计算表明:质子以羧基和苯环联合作桥迁移的路径为优势反应路径,裸反应的决速步吉布斯自由能垒为287.1 kJ·mol-1,2个水分子构成的链使决速步的吉布斯自由能垒降为144.9 kJ·mol-1。结果表明:布洛芬分子的手性转变存在多条可能的路径,水分子对布洛芬分子的H迁移异构反应有明显的催化作用,生命体内水分子的存在、温度的涨落、分子的频繁碰撞和某种酶的作用等综合因素,是导致左旋布洛芬旋光异构的原因。
手性;布洛芬;密度泛函理论;过渡态;微扰理论;吉布斯自由能
布洛芬(Ibu)的分子式是C13H18O2,根据其构型和旋光性的不同,可分为左旋布洛芬(SIbu)和右旋布洛芬(RIbu)。其具有较好的抗炎、镇痛和解热作用,临床上广泛用于治疗风湿性关节炎、强直性脊椎炎和神经炎等疾病。由于Ibu的重要应用,学者们对它进行了广泛的研究。文献[1]研究了有机溶剂中布洛芬的脂肪酶催化对映选择性酯化。文献[2]进行了外消旋布洛芬的动力学拆分模拟和实验的研究。文献[3]的研究发现,RIbu具有较好的药理活性,其疗效和安全性都优于外消旋Ibu。文献[4-5]的研究发现,RIbu的活性是SIbu的160倍,是外消旋体的1.6倍,SIbu在生命体内可以缓慢实现旋光异构。
文献[6]的研究发现,孤立条件下Ibu分子的手性转变反应路径是羧基内H迁移后,手性碳上的氢以新羰基氧为桥转移到手性碳的另一侧,完成手性转变。文献[7]的研究发现,Ibu在水环境下的手性转变有2条路径,分别是手性碳上的氢以水分子为媒介只以羰基氧为桥,羧基内H迁移后手性碳上的氢再以水分子为媒介以新羰基氧为桥。文献[8]研究了扶手椅型单壁碳纳米管对Ibu分子手性转变的限域影响,结果表明,随着纳米管尺寸的减小,反应路径由2条变为1条,决速步能垒逐渐变小。
目前市场销售的Ibu多数为消旋体[9],原因是利用不对称合成方法获得光学纯RIbu的成本很高,利用拆分方法获得RIbu的同时会产生几乎同等数量的“劣构体”SIbu。寻找有效的使SIbu转化为RIbu的途径显得极其重要,这就需要对Ibu手性转变机理进行深入地研究。本工作对Ibu分子手性转变的裸反应机理和水分子对决速步基元反应的催化作用进行了全面而细致地研究,较好地说明了SIbu在生命体内可以缓慢地向RIbu转变,对进一步研究复杂环境下Ibu的手性转变具有及其重要的意义。
1 计算方法
利用密度泛函理论的B3LYP方法[10-11],结合6-31+G(d,p)基组,全优化SIbu向RIbu转变过程中的各个驻点。通过对过渡态[12-14]虚频振动模式的分析和内禀反应坐标(IRC)[15-16]计算,对过渡态进行确认。采用微扰理论的MP2方法[17-18],在MP2/ 6-311++G(2df, pd)理论水平,计算体系的单点能。进行吉布斯自由能热校正,利用Gtotal=ESP+G(其中,Gtotal为总自由能,ESP为高水平的单点能,G为吉布斯自由能热校正)计算总自由能,最后绘制出反应过程自由能势能面。文中计算均由Gaussian09程序包完成[19]。文中大的最深色球为C原子,大球为O原子,小球为H原子。
2 结果与讨论
单体S型和R型Ibu的几何构型[6],见图1(a)和(b)。

图1 S型与R型布洛芬分子的几何结构Fig.1 Geometries of S and R type ibuprofen molecules
通过对图1的分析与研究发现,单体Ibu从S型向R型异构反应通道总体可以分为三个,一个是手性碳上的质子以羰基氧为桥迁移,实现手性转变,称之为a通道;第二个是羧基内先实现质子迁移,形成新的羰基,而后手性碳上的质子以新的羰基氧和苯环为桥迁移,实现手性转变,称之为b通道。第三个是羧基内先实现质子迁移后,手性碳上的质子只以新的羰基氧为桥迁移,实现手性转变,此工作文献[6]已有研究。下面对a和b通道分别进行讨论。2.1SIbu在a通道不同路径上的手性转变反应机理
对于SIbu在a通道的手性转变反应,又可以分为两条路径,一个是手性碳上的质子直接迁移到羰基氧,我们称之为a1路径;另一个是手性碳上的质子向手性碳上的甲基与甲基上的质子向羰基氧的双质子协同迁移,我们称之为a2路径。
SIbu在a1路径的反应历程见图2,首先SIbu经过32H向羰基氧31O迁移的过渡态a1TS1,异构为中间体a1INT1,其羧基是质子化的。此基元反应过程中,SIbu的32H-12C键长为0.109 46 nm,a1TS1的32H和12C之间的距离为0.156 37 nm,即从SIbu到过渡态a1TS1是32H和12C断键的过程。而SIbu的32H-12C键的结合能是较大的,因此从SIbu到过渡态a1TS1需越过较大的能垒。中间体a1INT1的二面角12C-16C-17C-31O和12C-17C-30O-31O分别为-1.298°和-179.710°,说明骨架12C、16C、17C、30O、31O基本共面。二面角12C-17C-31O-32H为-9.488°,说明32H偏向纸面里侧,此构型有利于32H在纸面里侧向手性碳迁移。
a1INT1之后的过程,又分为两个分路径am和an。am是a1INT1的质子化羧基进一步异构后,32H直接向手性碳的迁移;an是32H向16C和16C上在纸面里侧的质子在纸面里向手性碳的双质子协同迁移。对于分路径am,a1INT1经过32H和33H在纸面里外翻转的过渡态amTS2,异构成32H和33H分别在纸面里侧和外侧的中间体amINT2。此基元反应的过渡态无断键,反应能垒不会很高。amINT2的二面角30O-17C-31O-32H为50.946°,12C-17C-31O-32H为128.566°,因此,amINT2的构型更有利于32H在纸面里向手性碳迁移。接下来amINT2经过amTS3,实现32H在纸面里向手性碳迁移,形成此路径上的产物amP_R_Ibu,完成手性转变。此基元反应雷同于第1步骤SIbu→a1TS1→a1INT1,不再详细讨论。对于分路径an,a1INT1经过32H向16C和24H在纸面里侧向12C的双质子协同迁移的过渡态anTS2,异构成产物amP_R_Ibu,完成手性转变。此基元反应a1INT1的32H-31O键长为0.096 58 nm,16C-24H键长为0.109 92 nm,anTS2的32H和31O之间的距离为0.105 14 nm,16C和24H之间的距离为0.144 27 nm,从a1INT1到anTS2过程,32H和31O以及16C和24H均由成键变成断键。因此,从a1INT1到anTS2需要较大的能量,亦即anTS2会产生很高的能垒。
SIbu在a2路径的第1个基元反应的过渡态和中间体见图2,是SIbu先经过32H从手性碳12C向甲基的16C和甲基的26H向羰基氧31O的双质子协同迁移的过渡态a2TS1,异构成中间体产物a2INT1。此基元反应与前面的a1INT1→anTS2→amP_R_Ibu相似,anTS2有2个断键。但是由于SIbu→a2TS1→a2INT1需要断的键是12C-32H和16C-26H,12C-32H断裂需要的能量要大于32H-31O断裂需要的能量,因此,此基元反应所需克服的能垒高于a1INT1→anTS2→amP_R_Ibu的能垒,后面的计算说明了这一点。计算表明a2INT1全同于a1INT1,a2INT1以后的过程同于a1INT1后面的amTS2和anTS2之后的过程,这里不再赘述。
优化的反应路径上驻点几何结构见图2,计算的过渡态在虚频下的振动模式亦见图2。对过渡态在虚频下的振动模式进行的分析和对过渡态进行的IRC计算,确认了各个过渡态的可靠性。计算的各个驻点的吉布斯自由能热校正、单点能和过渡态的虚频见表1,吉布斯自由能热校正后的总自由能以及以SIbu为自由能零点的相对总自由能亦见表1。
根据表1的数据,绘制了SIbu在a通道不同路径手性转变反应过程的吉布斯自由能势能面示意图见图3。
从图3可以看出,SIbu在a通道4个不同路径的手性转变反应过程中,a1路径与am分路径构成的路径:SIbu→a1__TS1→a1_INT1→am_TS2→am_INT2→am_TS3→am_P_RIbu为优势反应路径,决速步能垒为295.8 kJ·mol-1,是由质子从手性碳向羰基迁移的过渡态a1_TS1产生的。优势反应路径的决速步能垒如此之高,说明孤立环境下的布洛芬不会在此通道发生异构。a2路径和am与an分路径构成的两个路径具有相同的决速步能垒422.1 kJ·mol-1,如此高的能垒,在一般的催化剂作用下也难以逾越。这说明单体布洛芬在a通道的手性转变极难进行。

图2 SIbu在a通道不同路径的手性转变过程Fig.2 Chiral transition process of SIbu in different paths of channel a
表1SIbu在a通道手性转变过程中,各驻点的吉布斯自由能热校正、过渡态的虚频、单点能、总吉布斯自由能及相对总吉布斯自由能
Table 1 Thermal recalibration Gibbs free energy, transition state imaginary frequency, single point energies, total Gibbs free energies, relative total Gibbs free energies of the each stationary point in channel a of chiral transition process ofSIbu
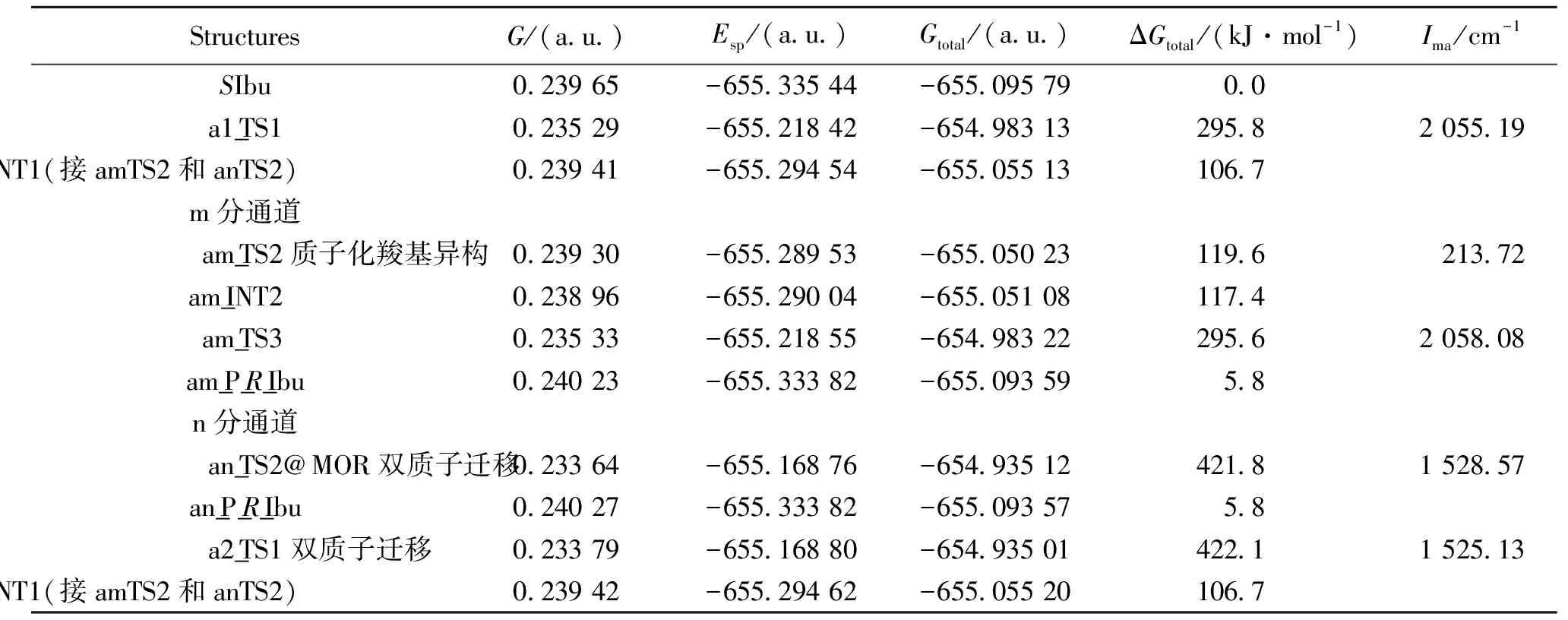
StructuresG/(a u )Esp/(a u )Gtotal/(a u )ΔGtotal/(kJ·mol-1)Ima/cm-1SIbu0 23965-655 33544-655 095790 0a1_TS10 23529-655 21842-654 98313295 82055 19a1_INT1(接amTS2和anTS2)0 23941-655 29454-655 05513106 7m分通道am_TS2质子化羧基异构0 23930-655 28953-655 05023119 6213 72am_INT20 23896-655 29004-655 05108117 4am_TS30 23533-655 21855-654 98322295 62058 08am_P_R_Ibu0 24023-655 33382-655 093595 8n分通道an_TS2@MOR双质子迁移0 23364-655 16876-654 93512421 81528 57an_P_R_Ibu0 24027-655 33382-655 093575 8a2_TS1双质子迁移0 23379-655 16880-654 93501422 11525 13a2_INT1(接amTS2和anTS2)0 23942-655 29462-655 05520106 7
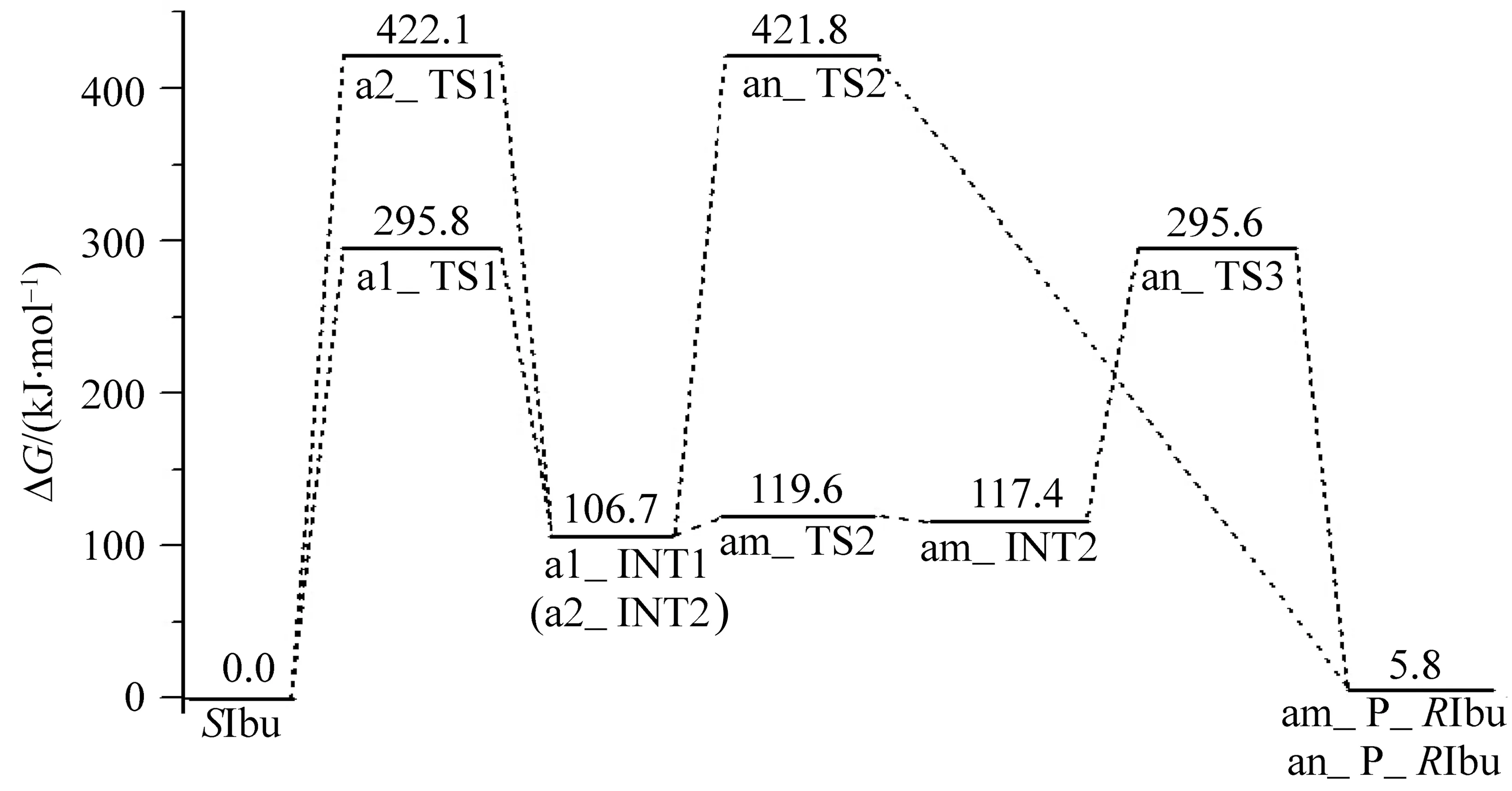
图3 SIbu在a通道不同路径手性转变反应的吉布斯自由能势能面示意图Fig.3 Gibbs free potential energy surfaces diagram of chiral transition process of SIbu in the different paths of channel a
2.2 SIbu在b通道不同路径上的手性转变反应机理
SIbu在b通道的手性转变反应过程见图4,SIbu到bINT2的过程文献[6]已有讨论,这里从略。bINT2经过32H从30O向5C迁移的过渡态bTS3异构成中间体产物bINT3。此基元反应bINT2的32H-30O键长是0.097 02 nm,bTS3的32H-30O间距是0.130 88 nm,32H和30O断键,因此,bTS3产生的能垒不会太低。从图4可以看出,bTS2是碳氢键12C-32H断裂,因此bTS3产生的能垒不会高于bTS2的能垒,后面的计算说明了这一点。

图4 SIbu在b通道不同路径的手性转变过程Fig.4 Chiral transition process of SIbu in different paths of channel b
从图4可以看出,bINT3之后又分为两个分路径b1和b2。b1路径是bINT3的9H经过过渡态b1TS4实现了从5C向30O的迁移,异构成为b1INT4。b1INT4的二面角12C-17C-30O-9H是9.569°,说明了9H偏向纸面里侧,其在纸面里侧向12C迁移的几率增加。b1INT4经9H向12C迁移的过渡态b1TS5异构成此路径的产物1,记作b1_P1_RIbu,完成手性转变。b1TS5的9C和30O的距离是0.126 61 nm,9C和30O是断键,因此会产生较高的能垒。b1_P1_RIbu又可以经过羧基上质子回迁的过渡态b1TS6,异构为此路径的产物2,记作b1_P2_RIbu。计算表明,b1TS6产生的对于b1_P1_RIbu和b1_P2_RIbu相互异构的能垒不太高,并且b1_P1_RIbu和b1_P2_RIbu的能量相差无几,因此,此路径是b1_P1_RIbu和b1_P2_RIbu两种产物共存。
b2路径是bINT3的9H经过过渡态b2TS4在纸面里直接迁移到12C,异构成产物b2_P1_RIbu,完成手性转变。bINT3的5C-9H键长是0.109 97 nm,b2TS4的5C和9H的距离是0.160 26 nm,5C和9H已经断键,因此,b2TS4会产生较高的能垒。计算表明,b2_P1_RIbu构型全同于b1_P1_RIbu,其也可经过b2TS5(计算表明全同于b1TS6)异构成产物b2_P2_RIbu。
全优化反应路径上的驻点,计算高水平的单点能。得到的各驻点几何结构及过渡态在虚频下的振动模式见图4,通过对过渡态进行虚频下的振动模式分析和IRC计算,确认了诸过渡态的可靠性。吉布斯自由能热校正、高水平的单点能和过渡态的虚频值见表2,吉布斯自由能热校正后的总自由能以及以SIbu为自由能零点的各驻点相对自由能亦见表2。
依据表2的数据,绘制了SIbu在b通道实现手性转变反应过程的势能面示意图,见图5。
从图5可以看出,SIbu在b-b1和b-b2两个分路径具有相同的决速步,决速步能垒是手性碳上的质子32H向羰基氧30O迁移的同一个过渡态b_TS2产生的,能垒是287.1 kJ·mol-1。结合2.1的图3可知SIbu在b通道的手性转变反应略占优势。b2_TS4和b1_TS5的能垒相差约为4.0 kJ·mol-1,可以认为基本相同,因此两个分路径b1和b2基本上无优劣之分。287.1 kJ·mol-1的能垒在常温下是难以逾越的,说明通常孤立环境下的布洛芬不会在b通道发生旋光异构。再结合2.1的结果得到结论:孤立环境下的布洛芬极具稳定性,不会发生旋光异构。
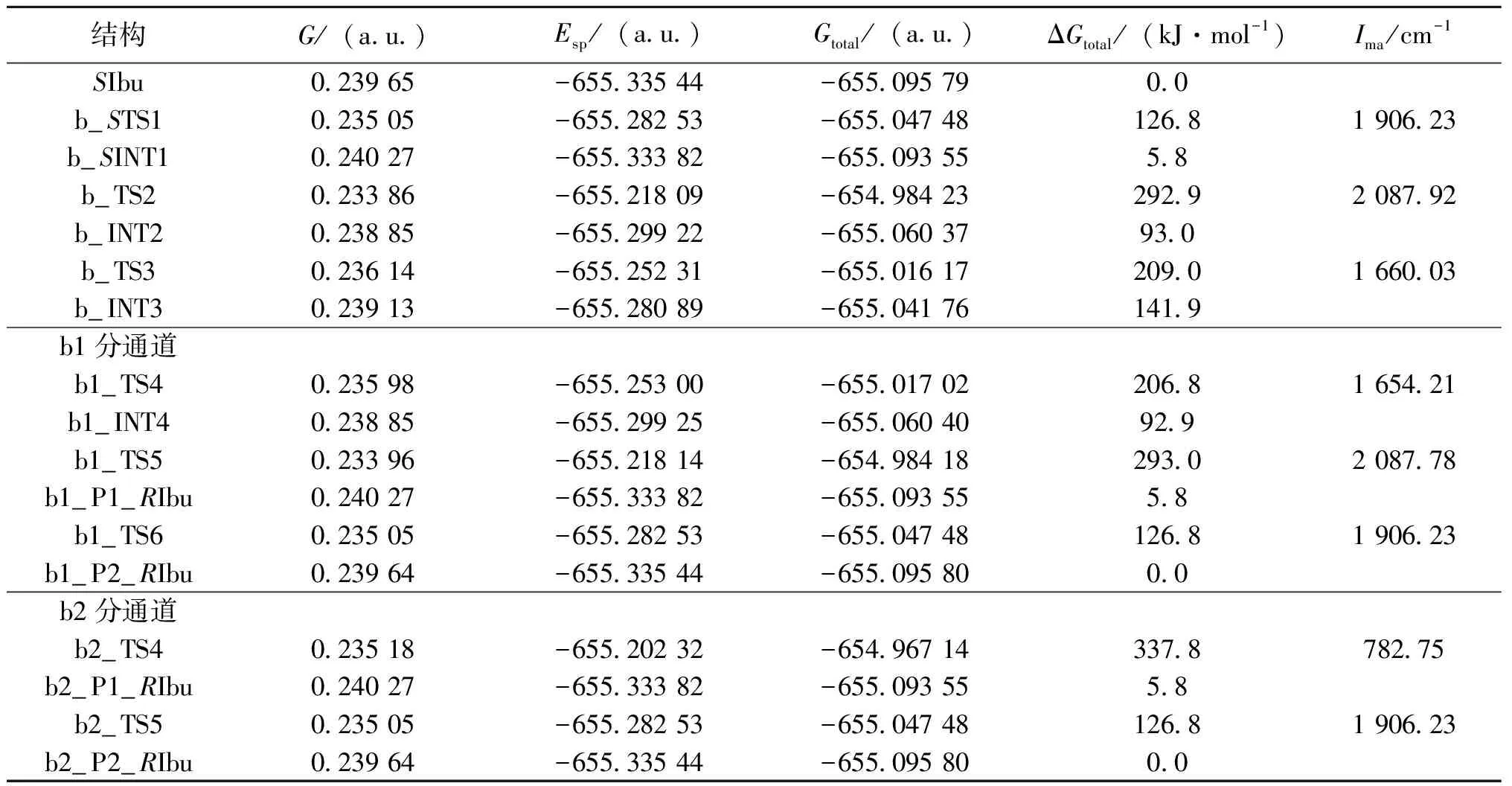
表2 SIbu在b通道手性转变过程中,各驻点的吉布斯自由能热校正、过渡态的虚频、单点能、总吉布斯自由能及相对总吉布斯自由能

图5 SIbu在b通道不同路径手性转变反应的吉布斯自由能势能面示意图Fig.5 Gibbs free potential energy surfaces diagram of chiral transition process of SIbu in the different paths of channel b
2.3 水助SIbu手性转变过程决速步骤基元反应机理
已有研究表明,水分子对H迁移反应具有很好的催化作用, 3个水分子构成的链的催化作用最好[20-24]。篇幅所限,对具有优势的b通道,分别讨论1个水分子、2个和3个水分子构成的链,催化bINT1→bTS2→bINT2的过程,以获得不同数目的水分子具有不同催化作用的机理。对于a通道,只给出3个水分子构成的链,催化SIbu→aTS1→aINT1的计算结果。
3个水分子构成的链,催化SIbu→aTS1→aINT1的反应历程,见图6 A,1个水分子、2个和3个水分子构成的链,催化b_INT1→b_TS2→b_INT2的反应历程,见图6 B。SIbu与32H和31O前面的3个水分子以氢键结合,形成水合分子SIbu·3H2O,经过渡态a_TS1·3H2O,异构为aINT1·3H2O,实现了3个水分子助SIbu→aTS1→aINT1的反应过程,质子从手性碳转移到羧基。b_INT1与32H和30O前面的1个、2个和3个水分子以氢键结合,形成的水合分子记作b_INT1·1H2O、b_INT1·2H2O和b_INT1·3H2O,它们经过渡态b_TS2·1H2O、b_TS2·2H2O和b_TS2·3H2O,实现了水助b_INT1→b_TS2→b_INT2的反应过程,质子从手性碳转移到羧基。
计算的水助SIbu手性转变过程决速步骤各个驻点的结构和过渡态在虚频下的振动模式见图6,吉布斯自由能热校正、单点能及过渡态的虚频见表3,吉布斯自由能热校正后的总自由能及相对总自由能亦见表3。通过优化各个过渡态沿其虚频振动的两个方向调节得到的结构,得到了每个过渡态对应的反应物和产物,验证了诸过渡态的可靠性,对过渡态进行的IRC计算,进一步确认了过渡态。
依据表3的数据,绘制了水助SIbu手性转变过程决速步骤基元反应过程的吉布斯自由能势能面,见图7。
从图7可以看出,水分子作用下SIbu分子的手性转变仍然是反应通道b略具优势,2个水分子构成的链助H迁移反应,决速步能垒基本降到最小,大小为144.9 kJ·mol-1,比前面裸反应的决速步能垒287.10 kJ·mol-1降低近50%。3个水分子构成的链助H迁移反应,相对于2个水分子构成的链助H迁移的反应能垒几乎没有下降。1个水分子助H迁移反应,决速步能垒为182.4 kJ·mol-1,说明1个水分子助H迁移反应的催化作用明显减弱。对于144.9 kJ·mol-1的能垒,若考虑到生命体内温度的涨落、分子的频繁碰撞和某种酶的作用是有越过的几率存在的。因此,SIbu在生命体内可以缓慢地旋光异构,向RIbu转化。
为进一步理解水分子数目对布洛芬分子手性转变的影响,将环形过渡态b_TS2·1H2O、b_TS2·2H2O、b_TS2·3H2O和a_TS1·3H2O的主要几何参数列于表4。

图6 水助SIbu手性转变过程决速步骤基元反应过程Fig.6 Elementary reaction process of rate-determining step of water-assisted SIbu chiral transition
表3 水助SIbu手性转变过程决速步骤基元反应各驻点的吉布斯自由能热校正、过渡态的虚频、单点能、总自由能及相对总自由能
Table 3 Thermal recalibration Gibbs free energy, transition state imaginary frequency, single point energies, total Gibbs free energies, relative total Gibbs free energies of the each stationary points in elementary reaction process of rate-determining step of water-assistedSIbu chiral transition

结构G/(a u )Esp/(a u )Gtotal/(a u )ΔGtotal/(kJ·mol-1)Ima/cm-1b_INT1·1H2O0 26014-731 66191-731 401770 0b_TS2·1H2O0 25662-731 58892-731 33230182 41693 91b_INT2·1H2O0 26129-731 60229-731 34100159 6b_INT1·2H2O0 28055-807 99189-807 711340 0b_TS2·2H2O0 27876-807 93491-807 65615144 91241 33b_INT2·2H2O0 28179-807 96366-807 6818777 4b_INT1·3H2O0 30177-884 32541-884 023640 0b_TS2·3H2O0 29967-884 26864-883 96897143 5962 62b_INT2·3H2O0 30405-884 29665-883 9926081 5SIbu·3H2O0 30051-884 32306-884 022550 0a_TS1·3H2O0 29945-884 26308-883 96363154 7809 38a_INT1·3H2O0 30195-884 29266-883 9907183 6

图7 水助SIbu手性转变过程决速步骤基元反应的吉布斯自由能势能面示意图Fig.7 Gibbs free potential energy surfaces diagram of elementary reaction process of rate-determining step of water-assisted SIbu chiral transition
从表4可以看出:8元环过渡态bTS2·2H2O和10元环过渡态bTS2·3H2O的键角12C-32H-34O和37O-39H-30O都远大于6元环过渡态bTS2·1H2O的键角12C-32H-34O和34O-35H-30O,并且接近180°,因此,bTS2·2H2O和bTS2·3H2O比bTS2·1H2O的结构稳定,产生的能垒低。从表4还可看出:10元环过渡态bTS2·3H2O的键角12C-32H-34O和37O-39H-30O比8元环过渡态bTS2·2H2O的大,更接近180°,对应的氢键强,似乎b_TS2·3H2O产生的能垒要明显低于bTS2·2H2O。但表4后两列二面角的数据反映出,10元环过渡态bTS2·3H2O与8元环过渡态bTS2·2H2O相比较,明显偏离平面,这又使得bTS2·3H2O比8元环过渡态bTS2·2H2O不稳定。因此,综合看来bTS2·3H2O与bTS2·2H2O产生的能垒不会相差很大。从表4还可看出:a_TS1·3H2O环形过渡态的氢键键角虽然较大,但环形过渡态结构严重偏离平面,造成过渡态结构张力大,不稳定,产生的能垒相对增加。这从过渡态的结构特性上说明了图7的合理性。

表4 b_TS2·1H2O、b_TS2·2H2O、b_TS2·3H2O和a_TS1·3H2O的主要结构参数
3 结论与展望
水助决速步骤过渡态结构的分析表明:水助质子从手性碳向羰基迁移过程的8元环过渡态bTS2·2H2O和10元环过渡态bTS2·3H2O对应的氢键键角都远大于6元环过渡态bTS2·1H2O,bTS2·3H2O和a_TS1·3H2O的氢键键角更趋于180°;过渡态bTS2·2H2O的8元环结构基本共面;过渡态a_TS1·3H2O和bTS2·3H2O的10元环结构明显偏离平面。反应路径研究发现:标题反应有2个通道,分为6条路径,分别是质子H只以羰基氧、依次以甲基碳和羰基O和以羧基和苯环联合作桥,从手性C的一侧迁移到另一侧。势能面计算表明:质子以羧基和苯环联合作桥迁移的路径为优势反应路径,裸反应的决速步吉布斯自由能垒为287.10 kJ·mol-1,2个水分子构成的链使决速步的吉布斯自由能垒降为144.9 kJ·mol-1。通过考察布洛芬分子手性转变的多条可能反应路径,发现水分子对布洛芬分子的H迁移异构反应有明显的催化作用。生命体内水分子的存在、温度的涨落、分子的频繁碰撞和某种酶的作用等综合因素,是导致SIbu旋光异构的原因。
纳米管等纳米孔道材料对布洛芬手性转变的限域催化,分子筛等绿色环保材料对布洛芬手性转变的择形、限域催化和酸催化更具实际意义,相关的研究正在进行中。
[1] DUCRET A, TRANI M, LORTIE R. Lipase-catalyzed enantioselective esterification of ibuprofen in organic solvents under controlled water activity[J]. Enzyme and Microbial Technology, 1998, 22(4): 212-216.
[2] SUBHASH B, WEI S L. Enzymatic membrane reactor for the kinetic resolution of racemic ibuprofen ester: modeling and experimental studies [J]. Chemical Engineering Science,2004,59(22/23): 5061-5068.
[3] 肖方清. 右旋布洛芬的制备[J]. 中国医药工业杂志, 2000, 31(11):486-488.
[4] 林文辉.手性药物布洛芬的体内药物动力学研究[D].沈阳药科大学,2004:8-10.
[5] CHENG H, ROGERS J D, DEMETRIADES J L, et al. Pharmacokinetics and bioinversion of ibuprofen enantiomers in humans [J]. Pharmaceutical Research, 1994, 11(6): 824-830.
[6] 邹晓威,梅泽民,王佐成,等.孤立条件下布洛芬分子手性转变过程的理论研究[J].原子与分子物理学报,2015, 32(2):173-180.
[7] 梅泽民, 王佐成,闫红彦,等.水环境下布洛芬分子的手性转变机理[J].吉林大学学报(理学版), 2015,53 (2):331-339.
[8] 王佐成,梅泽民,吕洋,等. 扶手椅型单壁碳纳米管的尺寸对布洛芬分子手性转变的限域影响[J]. 复旦学报(自然科学版)2015,54(2): 234-244.
[9] 赵亚华. 分子生物学教程[M].北京:科学出版社,2011: 5-6.
[10] BECKE A D. Density-functional thermochemistry. III. The role of exact exchange [J]. Chem Phys, 1993, 98(7): 5648-5652.
[11] PARR R G, YANG W. Density-functional theory of atoms and molecules[M]. Oxford: Oxford University Press, 1994.
[12] EYRING H. The activated complex and the absolute rate of chemical reaction [J]. Chemical Reviews, 1935, 17(1): 65-77.
[13] GARRETT B C, TRUHLAR D G. Generalized transition state theory. Classical mechanical theory and applications to collinear reactions of hydrogen molecules [J]. Journal of Physical Chemistry, 1979, 83(8): 1052-1079.
[14] GARRETT B C, TRUHLAR D G. Criterion of minimum state density in the transition state theory of bimolecular reactions [J]. The Journal of Chemical Physics, 1979, 70(4): 1593-1598.
[15] GONZALEZ C, SCHLEGEL H. Reaction path following in mass-weighted internal coordinates [J]. Journal of Physical Chemistry, 1990, 94(14): 5523-5527.
[16] ISHIDA K, MOROKUMA K, KOMORNICKI A. The intrinsic reaction coordinate. Anabinitiocalculation for HNC→HCN and H-+ CH4 →CH4+ H-* [J]. The Journal of Chemical Physics, 1977, 66(5): 2153-2156.
[17] 徐光宪,黎乐民,王德民. 量子化学(中册)[M]. 北京:科学技术出版社,1985: 962-986.
[18] BINKLEY J S, POPLE J A. Moeller-Plesset theory for atomic ground state energies[J]. Int J Quantum Chem, 1975,9(2):229-236.
[19] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09. Revision D.01 [CP]. Pittsburgh USA: Gaussian Inc, Wallingford CT, 2013.
[20] TIAN C J,PENG X,WANG Z G,et al. Enantiomerization mechanism of thalidomied and the role of water and hydroxide Ions[J]. Chem Eur J,2012,18:14305-14313.
[21] 刘凤阁,吕洋,王佐成,等. 水环境下赖氨酸分子的手性转变机理[J].武汉大学学报(理学版), 2015,61(5): 491-496.
[22] 佟华,梅泽民,王佐成,等. α-Ala限域在螺手性SWBNNT(10,6)与水复合环境下的手性转变机理[J]. 复旦学报(自然科学版),2015,54(4): 529-540.
[23] 闫红彦,王佐成,佟华,等.缬氨酸分子的手性转变及水分子的催化机理[J]. 中山大学学报(自然科学版),2016,55(2): 68-75.
[24] 王彦全,王佐成, 闫红彦,等. 水环境下基于氨基作氢迁移桥梁α-丙氨酸的手性转变机制[J].中山大学学报(自然科学版), 2016,55(5):57-65.
Bare reaction mechanism of chiral transition of ibuprofen molecules and the catalysis of water molecules using carbonyl and benzene ring as H transfer bridge
GAOFeng1,WANGZuocheng1,YANHongyan2,YANGXiaocui1,TONGHua1
( 1. The college of Physics, Baicheng Normal College, Baicheng 137000, China;2.Computer Science Department, Baicheng Normal College, Baicheng 137000, China )
The bare reaction of chiral transition of ibuprofen molecules and the mechanism of water-assisted proton transfer from carbon to carbonyl were studied using the B3LYP method of density functional theory and the MP2 method of perturbation theory. The molecular structure analysis showed that the hydrogen bond angles corresponding to the eight membered ring transition state bTS2·2H2O, and the ten membered ring transition state bTS2·3H2O in the processes of water-assisted proton transfer from carbon to carbonyl were all much larger than that corresponding to the six membered ring transition state bTS2·1H2O. Moreover the eight membered ring structure of transition state bTS2·2H2O was almost coplanar, and the ten membered ring structure of transition state a_TS1·3H2O/bTS2·3H2O was obviously out of plane. The study on the reaction path showed that there were six paths in the title reaction, where respectively proton only using ketonic O or methyl C and carbonyl O or carbonyl and benzene ring as the transfer bridge from one side to the other of chiral C. Calculations of potential energy surface showed that the path using proton was the dominant reaction channel, and carbonyl and benzene ring as the transfer bridge was the dominant reaction path. The Gibbs free energy barrier of the rate-determining step of bare reaction was 287.1 kJ·mol-1, which would be reduced to 144.9 kJ·mol-1because of the chains constituted by two water molecules. The results showed that the chiral transition of ibuprofen molecules could be realized in multiple paths and the water molecules had a better catalysis on H transfer heterogeneous reaction of ibuprofen molecules, as well as the presence of water molecules in the body of a life, temperature fluctuations, molecular frequent collisions and the action of some enzyme conditions were the cause of S-Ibu optical isomers.
chiral; ibuprofen; density functional theory; transition state; perturbation theory; Gibbs free energy
10.13471/j.cnki.acta.snus.2016.06.018
2016-05-09
吉林省科技发展计划资助项目(20160101308JC)
高峰(1983年生),男;研究方向:原子与分子物理;通讯作者:王佐成;E-mail:wangzc188@163.com
O641.12
A
0529-6579(2016)06-0115-10
