湘葛一号根HPLC指纹图谱及5种异黄酮含量的同时测定
2016-05-28叶惠煊邹亲朋刘向前
戴 玲,叶惠煊,邹亲朋,李 芝,刘向前*
(1.长沙博海生物科技有限公司,长沙 410205;2.湖南中医药大学药学院,长沙 410208;3.九芝堂股份有限公司,长沙 410021)
湘葛一号根HPLC指纹图谱及5种异黄酮含量的同时测定
戴玲1,叶惠煊2,3,邹亲朋1,李芝2,刘向前2*
(1.长沙博海生物科技有限公司,长沙410205;2.湖南中医药大学药学院,长沙410208;3.九芝堂股份有限公司,长沙410021)
摘要:目的采用高效液相色谱法建立湘葛一号根指纹图谱,并同时测定5种异黄酮的含量。方法色谱条件:采用AT·LICHROM C18色谱柱(250 mm×4.6 mm,5 μm);以甲醇-水溶液为流动相进行梯度洗脱;流速为1.0 mL·min-1;检测波长为250 nm;柱温为30 ℃。对不同采收期的10批样品的图谱进行测定,运用中国药典委员会的“中药色谱指纹图谱相似度评价软件2004AB”进行评价,并对5种异黄酮成分进行含量测定。结果建立了湘葛一号根HPLC指纹图谱,各色谱峰分离度良好,确定了7个共有峰,并采用对照品指认了其中5个色谱峰。含量测定条件通过方法学验证,葛根素、大豆苷、染料木苷、大豆苷元、染料木素的线性范围分别为5.00~50.00,1.00~10.00,1.82~18.20,1.00~10.00和2.00~20.00 μg·mL-1,线性关系良好(r>0.999),平均加样回收率为98.8%~101.8%,5种异黄酮含量测定结果可初步确定适宜采收期为12月份。结论该方法所建立的湘葛一号根指纹图谱特征性强、方法简单、结果可靠,结合主要有效成分含量测定可更好地用于其质量控制,对其鉴定也具有参考价值。
关键词:异黄酮;湘葛一号;高效液相色谱法;指纹图谱
野葛Puerarialobata(Willd.)Ohwi和粉葛PuerariathomsoniiBenth的干燥根收载于历版《中国药典》,其富含一系列异黄酮类化合物,如大豆苷、葛根素、大豆苷元、染料木苷、染料木素等[1-5]。现代药理研究表明,葛根异黄酮具有降血压、抗氧化、降血糖、抗肿瘤等作用[6-10]。湘葛一号是我国第1个通过杂交选育的葛根新品种,因其具有品质好、产量高、抗性强、综合性状优良等优点,故在湖南多地大力推广栽培和应用。众所周知,药材由于采收期的不同,其有效成分的含量不同,发挥的作用也不同。目前,诸如小叶榕叶[11]、金钱草[12]、裸花紫珠[13]等药材,通过利用指纹图谱技术,标示其特征性的共有色谱峰图谱,能够较为全面地反映中药的内在质量,日益成为国内外广泛接受的中药质量评价模式[14-15]。湘葛一号根尚无质量控制标准研究,作者采用HPLC法建立了湘葛一号根的指纹图谱,考察了不同采收期湘葛一号根的质量差异,为湘葛一号根的采收及质量控制提供依据。
目前,大多数文献[16-17]采用同时测定葛根中的几种异黄酮含量来进行质量控制,但均需独立的对照品。且化学对照品大多来自天然产物,具有分离难度大或者单体不稳定等问题,这无疑增加了实际工作中的困难。一测多评法是只测定1种易得到成分从而实现对多个成分同步测定的方法,目前已成功运用于丹参[18]、预知子[19]、双青咽喉片[20]等中药及中成药的质量控制。本实验以葛根素为内标物,采用一测多评法建立其与大豆苷、染料木苷、大豆苷元和染料木素的校正因子,探讨了一测多评法测定湘葛一号根的多成分含量,为湘葛一号根的质量评价提供理论依据。
1仪器与材料
1.1仪器Agilent 1200高效液相色谱仪(美国Agilent公司);AUW220D分析天平(日本岛津公司);予华HH-S型水浴锅(巩义市英峪予华仪器厂);FW177中草药粉碎机(天津市泰斯特仪器有限公司)。
1.2材料葛根素、染料木苷、大豆苷、染料木素和大豆苷元均购于中国食品药品检定研究院,质量分数均≥98%,批号依次为110752-200511,111709-200501,111738-200501,111704-200501和111505-200402。湘葛一号根样品于2011年5~12月及2013年1月和9月采自湖南省常德市湘葛一号种植基地,由湖南天盛生物科技有限公司提供,经刘向前教授鉴定为豆科葛属植物PuerariathomsoniiBenth的干燥根,10批样品粉碎,过60目筛,备用。甲醇为色谱纯试剂;水为超纯水;其他试剂均为分析纯。
2指纹图谱方法与结果
2.1色谱条件色谱柱:AT·LICHROM C18色谱柱(250 mm×4.6 mm,5 μm);流动相:甲醇(A)-水(B),梯度洗脱,0~10 min,20%~25% A,10~15 min,25%~40% A,15~18 min,40%~46% A,18~25 min,46% A,25~30 min,46%~55% A,30~35 min,55%~65% A,35~45 min,65%~72% A,45~55 min,72%~20% A,55~60 min,20% A;体积流量:1 mL·min-1;柱温:30 ℃;检测波长:250 nm;进样量:10 μL。
2.2混合对照品溶液的制备称取对照品葛根素、大豆苷、大豆苷元、染料木素和染料木苷,精密称定,置于同一量瓶中,加适量甲醇,超声溶解,稀释至刻度,摇匀,制成质量浓度分别为50.00,10.00,18.20,10.00和20.00 μg·mL-1的混合对照品溶液,备用。
2.3供试品溶液的制备取批号201112粉末约1 g,精密称定,置于100 mL具塞锥形瓶中,精密加入50 mL体积分数为50%的乙醇,密塞,称定质量,在80 ℃水浴中加热回流50 min,放冷,再称定质量,加体积分数为50%的乙醇补足质量,摇匀,经0.22 μm微孔滤膜滤过,取续滤液,即得。
2.4方法学考察
2.4.1精密度实验精密吸取批号201112供试品溶液,按照2.1项下色谱条件,重复进样6次,记录各色谱图,计算共有峰相对峰面积和相对保留时间。结果显示,各共有峰的相对峰面积和相对保留时间的相对标准偏差(relative standard deviation,RSD)均小于3%,表明该仪器精密度良好。
2.4.2重复性实验取6份批号201112样品1 g,精密称定,按照2.3项下方法制备供试品溶液,按照2.1项下色谱条件测定,记录各色谱图,计算共有峰的相对峰面积和相对保留时间。结果显示,各共有峰相对峰面积和相对保留时间RSD值均小于3%,表明该方法重复性良好。
2.4.3稳定性实验取批号201112供试品溶液,放置于室温下,按照2.1项下色谱条件,分别于0,2,4,6,8,12和24 h进样,记录各色谱图,计算其共有峰相对峰面积和相对保留时间。结果显示,各共有峰相对峰面积和相对保留时间的RSD值均小于3%,表明供试品溶液在24 h内稳定。
2.5湘葛一号根指纹图谱共有模式的建立及相似度评价
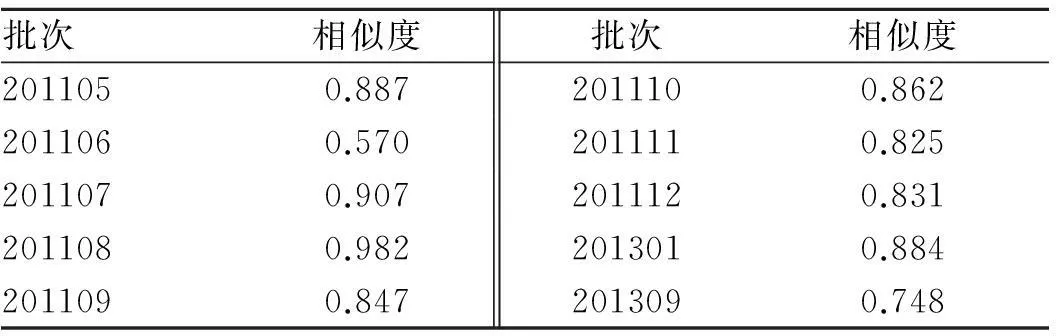
2.5.1不同采收期湘葛一号根指纹图谱的建立精密吸取2.3项下制备的10批次湘葛一号根供试品溶液各10 μL,依次注入高效液相色谱仪,记录色谱图时间65 min。采用《中药色谱指纹图谱相似度评价系统2004AB版》,将10批湘葛一号根的实验数据导入,设置参照图谱,匹配各色谱峰,生成对照图谱,见图1。葛根素是湘葛一号根中的特征性成分,故以其为参照峰,选取其中大于3%的7个共有峰作为指纹图谱的特征峰[21]。10批不同采收期湘葛一号根指纹图谱相似度计算结果见表1。
表1不同采收期湘葛一号根指纹图谱相似度计算结果
Tab.1 Similarity evaluation on ten batches of roots of Hu′nanPuerariathomsoniiBenth from different harvest periods
批次相似度批次相似度2011050.8872011100.8622011060.5702011110.8252011070.9072011120.8312011080.9822013010.8842011090.8472013090.748
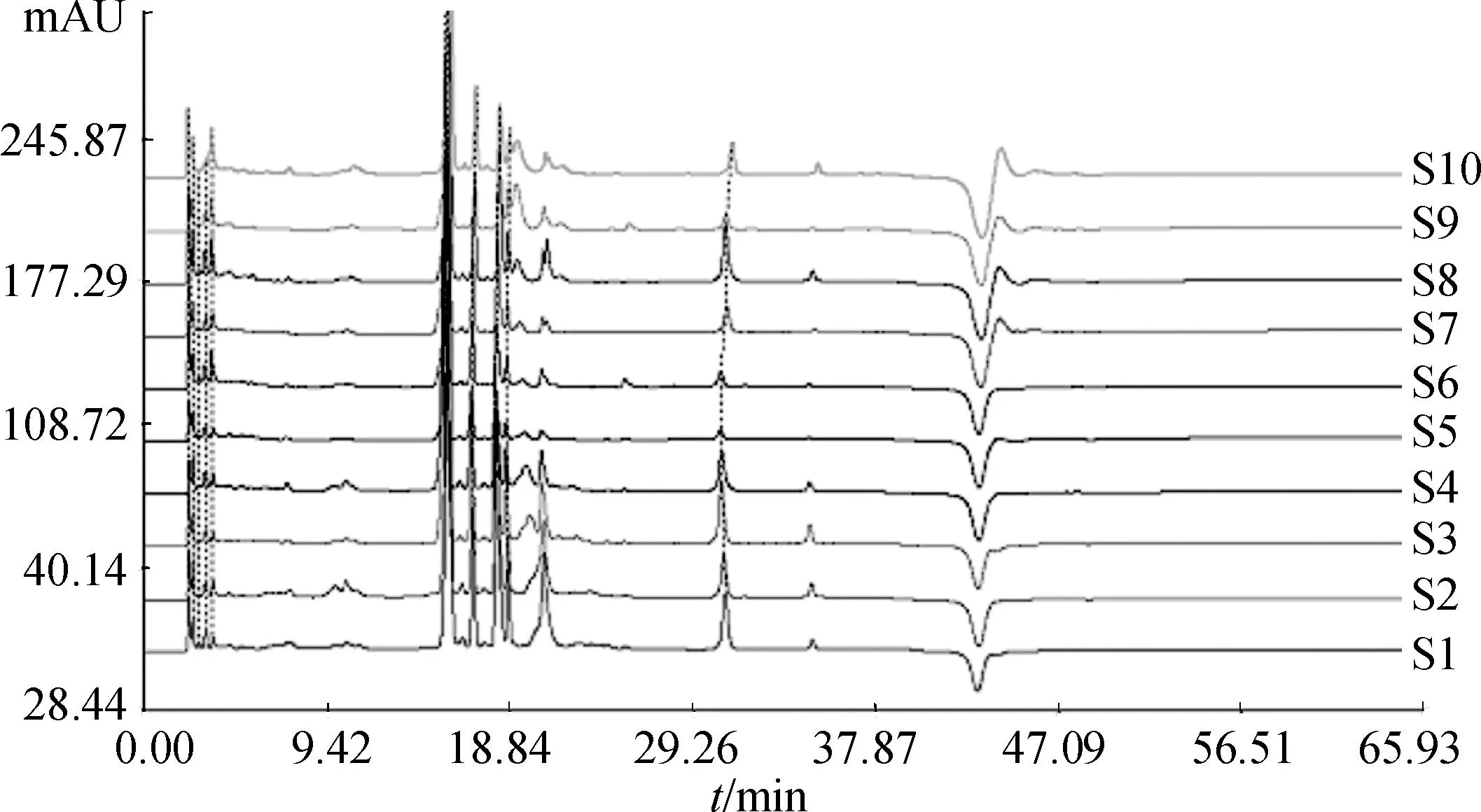
图1不同采收期湘葛一号根的HPLC叠加图谱
Fig.1 HPLC of the roots of Hu′nanPuerariathomsoniiBenth from different harvest periods
2.5.2共有峰的标定取2.2和2.3项下方法制备的混合对照品和供试品溶液各10 μL,分别注入高效液相色谱仪,记录各色谱峰。结果见图2。通过对不同采收期湘葛一号根的HPLC图进行比较,结果确定7个共有峰作为其指纹图谱的特征峰,并指认其中5个峰。
2.5.3不同采收期各有效成分峰面积的变化规律以2012年5~12月各批次样品5种异黄酮为指标的总峰面积计算,结果表明,5~12月样品总峰面积整体呈现先减后增的规律,其中5月份总量最高,9月份总量最低。

图2共有峰标定HPLC图
A.混合对照品;B.供试品;1.葛根素;2.大豆苷;3.染料木苷;4.大豆苷元;5.染料木素
Fig.2 Common peaks of HPLC chromatograms
A.mixed reference substances;B.roots;1.puerarin;2.daidzin;3.genistin;4.daidzein;5.genistein
3含量测定方法与结果
3.1色谱条件按照2.1项下色谱条件进行检测,以5种对照品计算理论板数大于3 000,分离度大于1.5。对照品溶液及供试品溶液的色谱图见图3。
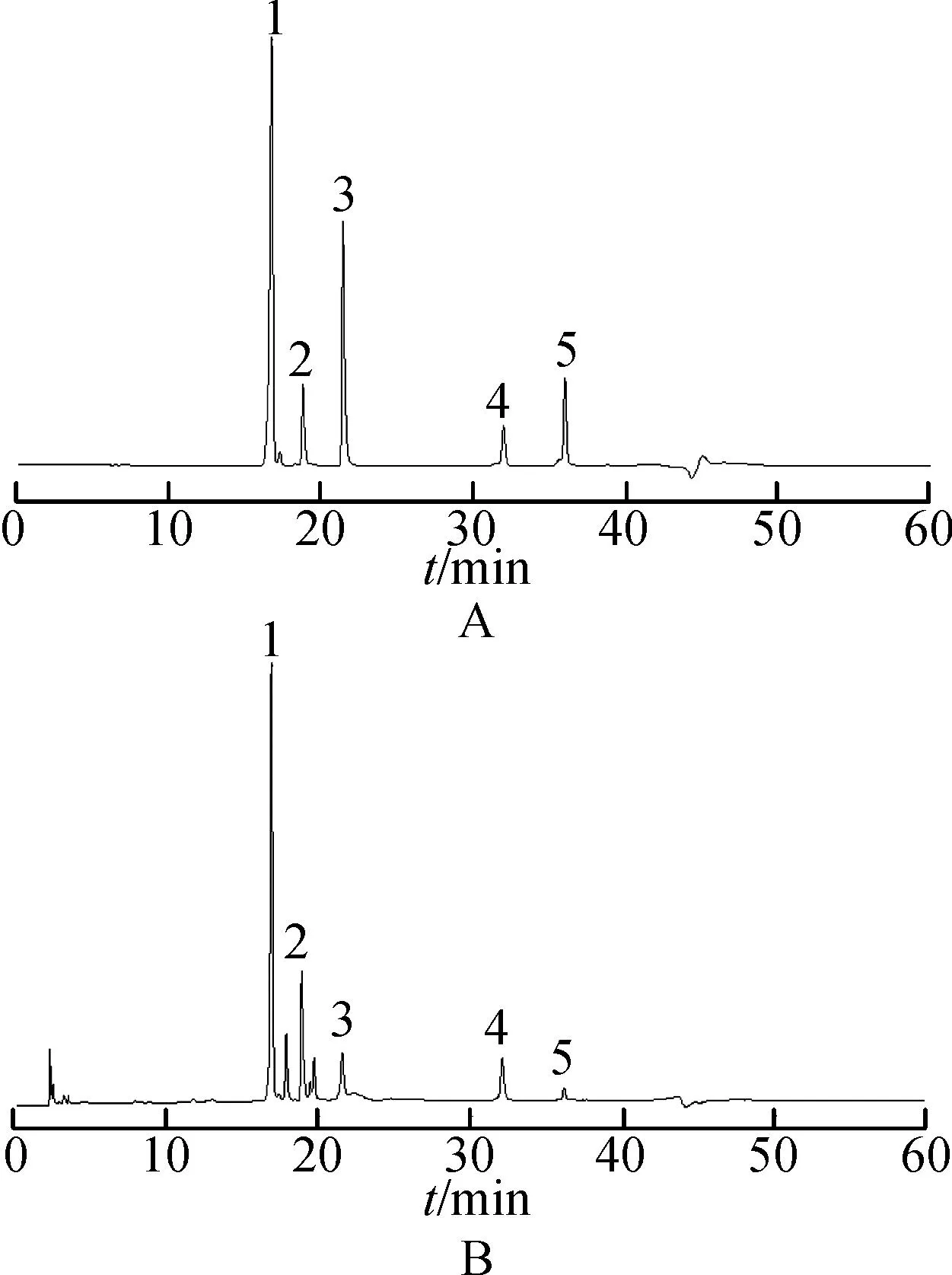
图3HPLC图
A.混合对照品;B.供试品;1.葛根素;2.大豆苷;3.染料木苷;4.大豆苷元;5.染料木素
Fig.3 HPLC chromatograms
A.mixed reference substances;B.roots;1.puerarin;2.daidzin;3.genistin;4.daidzein;5.genistein
3.2混合对照品溶液的制备同2.2项。
3.3供试品溶液的制备同2.3项。
3.4线性关系考察精密量取2.2项下混合对照品溶液适量,稀释到不同质量浓度,按照2.1项下色谱条件进样测定。以峰面积和混合对照品质量浓度分别为纵坐标和横坐标进行线性回归。分别按信噪比的3和10倍考察检测限和定量限,结果见表2。
表25种异黄酮成分的线性关系
Tab.2 Linear equations for five isoflavones

对照品回归方程r线性范围/μg·mL-1检测限/ng·L-1定量限/ng·L-1葛根素Y=2E+07X-1.35210.99945.00~50.000.942.85大豆苷Y=3E+06X+309.050.99921.00~10.000.240.73染料木苷Y=1E+06X+15.9290.99981.82~18.200.481.45大豆苷元Y=6E+06X-2739.40.99981.00~10.000.080.25染料木素Y=1E+07X+158500.99912.00~20.000.040.11
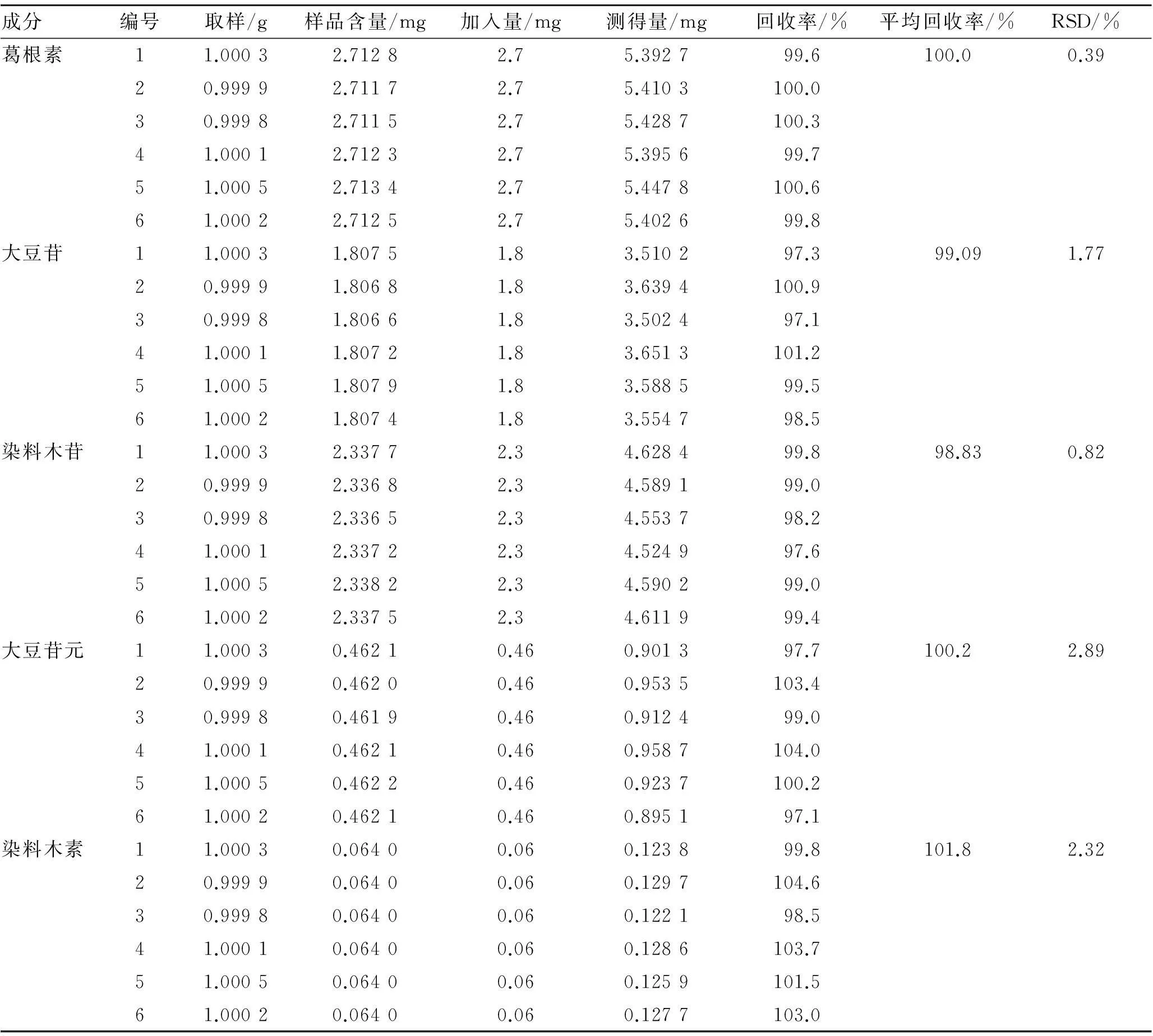
3.5加样回收实验取6份批号201112样品,每份1 g,精密称定,分别加入适量的混合对照品溶液(相当于葛根素2.70 mg、大豆苷1.80 mg、染料木苷2.30 mg、大豆苷元0.46 mg、染料木素0.06 mg),按照2.3项下方法制备供试品溶液,依次进样测定,计算各对照品的平均回收率。葛根素、大豆苷、染料木苷、大豆苷元和染料木素的平均回收率分别为100.0%,99.1%,98.8%, 100.2%和101.8%,RSD值分别为0.39%,1.77%,0.82%,2.89%和2.32%,见表3。
表3加样回收实验结果
Tab.3 Results of recovery test
(n=6)
3.6不同高效液相色谱仪和色谱柱的相对校正因子(RCF)重复性考察精密吸取2.2项下配制的混合对照品溶液,进样10 μL,平行重复6次,以葛根素为内标物,分别计算大豆苷、染料木苷、大豆苷元和染料木素的RCF值[22],取均值。实验考察了Agilent 1200型和Shimadazu LC-10AT型2种高效液相色谱仪及AT·LICHROM C18(250 mm×4.6 mm,5 μm)、Elite C18(250 mm×4.6 mm,5 μm)、Dikma C18(250 mm×4.6 mm,5 μm)3种色谱柱,结果表明,不同高效液相色谱仪和色谱柱对各成分的RCF值影响不大,结果见表4。
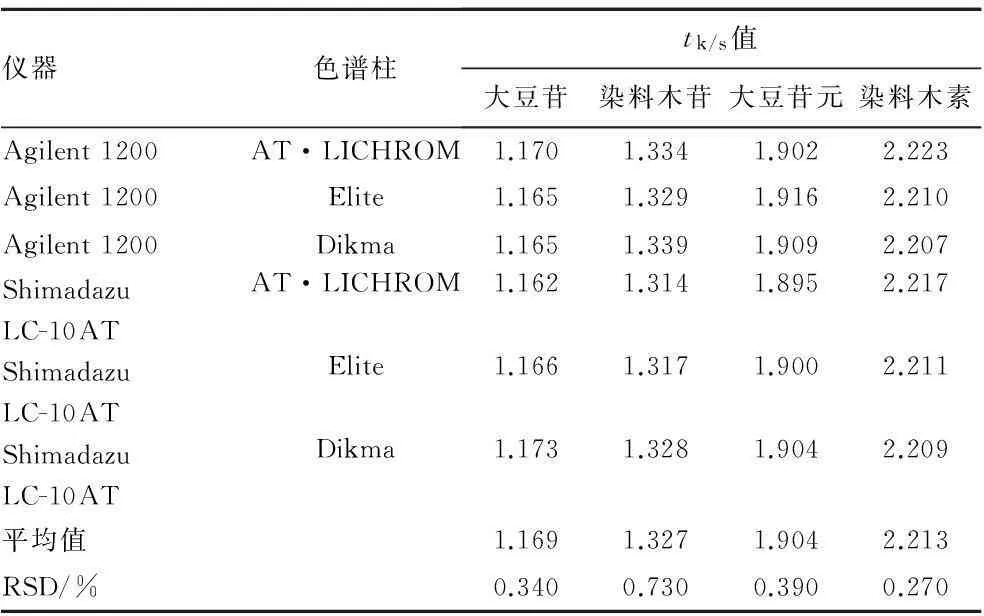
3.7 色谱峰定位参数考察由于本实验使用葛根素作为对照品,需要对大豆苷、染料木苷、大豆苷元和染料木素的色谱峰的位置进行定位,进而达到一测多评的目的。故利用相对保留时间(tk/s),即待测组分(tk)和内标物的保留时间(ts)之比:tk/s=tk/ts,可以定位待测组分。结果见表5。
表4不同仪器和色谱柱测定RCF值
Tab.4 RCF values of different instruments and chromatographic columns

(n=6)
表5不同仪器和色谱柱的tk/s值
Tab.5tk/svalues of different instruments and chromatographic columns
(n=6)
实验测得葛根素对大豆苷、染料木苷、大豆苷元和染料木素的相对保留时间(tk/s)的平均值分别为1.169,1.327,1.904和2.213。RSD在0.27%~0.73%之间。表明利用tk/s进行各色谱峰的定位是可行的。
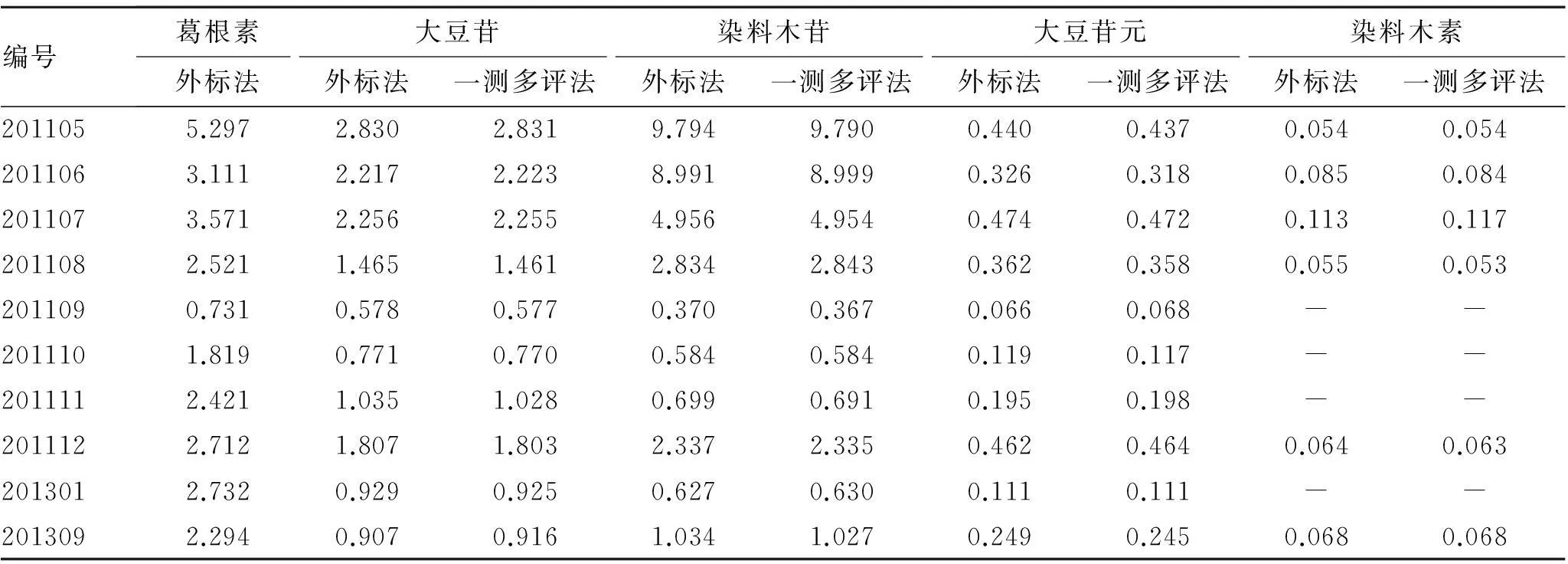
3.8样品含量测定分别精密吸取10批供试品溶液10 μL,进样测定。采用外标法和一测多评法分别计算葛根素、大豆苷、染料木苷、大豆苷元和染料木素的含量,结果显示无显著性差异。见表6。
4讨论
4.1色谱条件的选择本实验在前期研究工作的基础上[23],结合文献数据[24],结果显示,在250 nm波长处供试品溶液中色谱峰数目多且基线平稳,色谱信息量大,且各色谱峰分离度良好。通过对不同体积流量、不同柱温、不同进样量的色谱峰的数目及分离度比较,结果发现,最佳体积流量为1 mL·min-1,柱温为30 ℃,进样量为10 μL。
表6一测多评法与外标法测定湘葛一号根含量的结果比较
Tab.6 Comparison of quantitative determination results between single-maker and external standard method
(mg·g-1,n=3)
注:-表示未检测到。
4.2不同采收期湘葛一号根相似度评价在本实验条件下,指纹图谱中各色谱峰的分离度较好,得到了7个共有峰,并指认其中5个峰,结果全面反映了湘葛一号根的整体化学特征,体现其内在整体质量。实验结果采用均值法生成的对照图谱,通过相似度软件评价发现,仅有201107和201108两批样品的相似度大于0.9,说明不同采收期湘葛一号根所含成分及其含量差异较大,可能是不同的气候条件影响其有效成分的类别和含量。7个特征峰的相对保留时间的RSD均小于3%,符合指纹图谱的相关规定,但其相对峰面积相差很大,表明湘葛一号除了在固定产地的前提下,还需在适宜的采收期采收,才能确保湘葛一号根品质的一致性。
4.3湘葛一号最佳采收期的确定通过对不同采收期湘葛一号根的含量测定可知,均含有葛根素、大豆苷、染料木苷、大豆苷元和染料木素这5种异黄酮,且5种成分的含量在不同采收期呈现动态变化。葛根素、大豆苷和染料木苷含量均是5月份最高,9月份最低;大豆苷元和染料木素含量均是7月份最高,9月份最低。虽然湘葛一号根中5种成分总量在5月份相对较高,但是其传统采收期为秋冬季,此时根的产量最高,5种成分含量也不低。根据药用植物适宜采收期的确定原则,当药用植物的有效成分含量最高时期与产量最高时期不一致时,以两者乘积最大值的时期作为适宜采收期[25],因此将湘葛一号根的适宜采收期确定为12月。
4.4一测多评方法的探讨由于葛根素是葛属药材的主要药效成分,且在药材中含量相对较高,对照品稳定易得到。故本实验选用葛根素作为内标物,建立该成分与其他4种异黄酮类成分的RCF,从而实现一测多评。实验中常规外标法测得的含量与一测多评所测得的葛属药材中各成分含量间无显著性差异,说明一测多评法能够实现葛属药材的多成分含量的同步测定。同时,葛根异黄酮的RCF在湘葛一号不同部位及其他不同葛属品种的成功运用,为一测多评法推广到其余葛属药材及其不同部位的质量控制和评价提供了一定的依据。
5结论
本文利用指纹图谱研究及其相似度分析,并结合有效成分的含量测定,能够从整体上对湘葛一号进行质量评价,快速鉴别湘葛一号根药材及其最佳采收期,避免了仅仅采用1种或者几种有效成分含量的高低来片面地评价药材的优劣,为湘葛一号根的质量控制提供了一定的依据。
参考文献:
[1]Wu Yanfang,Wang Xinsheng,Fan Enguo. Optimisation of ultrasound-assisted extraction of puerarin and total isoflavones fromPuerariaelobataeradix (Puerarialobata(Wild.) Ohwi) with Response Surface Methodology [J]. Phytochem Anal, 2012,23(5): 513-519.
[2]Zhao Chenxi,Chan H Y,Yuan Dalin,et al. Rapid simultaneous determination of major isoflavones ofPuerarialobataand discriminative analysis of its geographical origins by principal component analysis[J]. Phytochem Anal,2011,22: 503-508.
[3]Yusakul G,Putalun W,Udomsin O,et al. Comparative analysis of the chemical constituents of two varieties ofPuerariacandollei[J]. Fitoterapia,2011,82(2): 203-207.
[4]Satoko S, Takuya K, Tsutomu I, et al. Quantitative analysis of miroestrol and kwakhurin for standardization of Thai miracle herb ‘Kwao Keur’ (Puerariamirifica) and establishment of simple isolation procedure for highly estrogenic miroestrol and deoxymiroestrol[J]. Nat Prod Res,2013, 45(27): 371-378.
[5]Sage R F,Coiner H A,Way D A, et al. Kudzu [Puerariamontana(Lour.) Merr.Variety lobata]: A new source of carbohydrate for bioethanol production [J]. Biomass Bioenergy,2009,33:57-61.

[7]Latiporn U,Thaweesak J,Waraporn P,et al. Down regulation of gene related sex hormone synthesis pathway in mouse testes by miroestrol and deoxymiroestrol [J]. Fitoterapia,2011,82:1185-1189.
[8]肖冰心,王倩,樊利青,等. 葛根黄酮提高桑白皮降糖活性及其机制研究[J].中国实验方剂学杂志,2013,19(3):179-183.
[9]Niu Ling, Li Dongye,Xia Yong, et al. Effect of puerarin on the myocardial perfusion and ventricular wall motion in patients with acute coronary syndrome [J]. J Geria Cardi,2008,5(2):50-53.
[10]Chang Y,Hsieh C Y,Peng Z A,et al. Neuroprotective mechanisms of puerarin in middle cerebral artery occlusion-induced brain infarction in rats [J]. J Biomed Sci,2009,16:9-21.
[11]黄洋,邵慧凯,路丽,等. 不同采收期小叶榕叶HPLC指纹图谱研究[J]. 中草药,2014,45(2):271-275.
[12]陈丰连,张文进,徐鸿华. 不同采收期及不同产地广金钱草地上部分HPLC指纹图谱研究[J]. 中国实验方剂学杂志, 2010,16(14):96-98.
[13]刘幼娴,谷陟欣,卢凤来,等.不同采收期裸花紫珠的HPLC指纹图谱研究[J].广西植物,2014,34(2):174-178.
[14]何晋浙,蒉霄芸,张安强,等.灵芝醇提生物活性物质的指纹图谱分析及质控评价[J]. 中草药,2011,42(6):1125-1129.
[15]Shen Z,Zhang W T,Hua Y F,et al. Fingerprint analysis of four variants ofChrysanthemiMorifoliFlosby RP-HPLC [J]. Chin Herb Med,2010,2(2): 153-156.
[16]刘喜舞,戴玲,叶惠煊,等. HPLC法同时测定中国、泰国葛根中异黄酮的含量[J].中国药师,2013,16(5): 668-670.
[17]裴香萍,刘亚明,刘计权,等. 野葛、粉葛与云南葛中葛根素、大豆苷、大豆苷元、染料木素的含量比较[J]. 中国药房,2012,23(47): 4462-4464.
[18]杨东亮,马贵芝,王娟,等. 一测多评法测定杜麻颗粒中5种成分的含量[J].西北药学杂志,2015,30(3):247-251.
[19]宋永贵,张武岗,刘岩庭,等. 一测多评法同时测定预知子中4种三萜皂苷[J]. 中草药,2012,43(7): 1418-1421.
[20]何兵,刘艳,杨世艳,等. HPLC一测多评法同时测定双青咽喉片中10种成分[J]. 中草药,2013,44(8): 974-981.
[21]宋丽明,王文燕,张智超. 陕西安康葛根药材的HPLC指纹图谱研究[J]. 中草药,2008,39(3):436-438.
[22]王智民,高慧敏,付雪涛, 等. “一测多评”法中药质量评价模式方法学研究[J]. 中国中药杂志,2006,31(23): 1925-1928.
[23]叶惠煊,刘向前,刘恒言,等. 湘葛一号总黄酮与气候因子的灰色关联度分析[J]. 西北药学杂志,2013,28(5): 443-447.
[24]尤春雪,张振秋,李峰,等. HPLC波长切换技术对葛根中8种成分的测定及指纹图谱研究[J].中草药,2013, 44(5):616-621.
[25]王立德,张兴翠,孙滢. 野葛主要成分积累动态变化研究[J]. 中国药学杂志,2008,43(13):974-977.
HPLC fingerprint for roots of Hu′nanPuerariathomsoniiBenth and simultaneous determination of five isoflavones
DAI Ling1, YE Huixuan2,3, ZOU Qinpeng1, LI Zhi2, LIU Xiangqian2*
(1. Changsha Broad-Ocean Bio-Science and Technique Limited Company, Changsha 410205, China 2.School of Pharmacy, Hu′nan University of Chinese Medicine, Changsha 410208, China; 3. Jiuzhitang Limited Company,Changsha 410021, China)
Abstract:ObjectiveAn HPLC fingerprint method for the roots of Hu′nan Pueraria thomsonii Benth in different harvest periods was developed,and quantities of five isoflavones were determined simultaneously. MethodsThe chromatography was performed on an AT·LICHROM C18column (250 mm×4.6 mm,5 μm) with a mobile phase of methanol-water and gradient elution. The flow rate was 1.0 mL·min-1. The detection wavelength was 250 nm. The column temperature was 30 ℃. The chromatograms of 10 batches of samples in different harvest period was determined by HPLC.ResultsIn the chromatographic fingerprint,7 peaks were selected as the characteristic peaks to assess the similarities of different samples according to similarity evaluation for chromatographic fingerprint of traditional Chinese medicine (2004AB). The chemical components contained in the roots of Hu′nan Pueraria thomsonii Benth were effectively separated. Five common fingerprint peaks were labeled from the seven common peaks. In quantitative analysis, the five components showed good regression(r>0.999), linear ranges of puerarin,daidzin,genistin, daidzein, genistein were 5.00-50.00,1.00-10.00,1.82-18.20,1.00-10.00 and 2.00-20.00 μg·mL-1,and their recoveries were in the range of 98.8%-101.8%,and the optimal harvest period was December.ConclusionThe combination of chromatographic fingerprint and quantitative analysis can be readily used as a quality control method for Hu′nan Pueraria thomsonii Benth.
Key words:isoflavone;Hu′nan Pueraria thomsonii Benth;HPLC;fingerprint
(收稿日期:2015-09-15)
中图分类号:R284
文献标志码:A
文章编号:1004-2407(2016)03-0239-06
doi:10.3969/j.issn.1004-2407.2016.03.006
*通信作者:刘向前,男,教授
作者简介:戴玲,女,硕士研究生
基金项目:长沙市科技局科技计划重点项目(编号:K1403122-31)
