异丁烷-丁烯固体酸烷基化反应动力学研究进展
2016-05-17刘晓宇周祥郭锦标王鑫磊付翁中国石油化工股份有限公司石油化工科学研究院北京100083
刘晓宇,周祥,郭锦标,王鑫磊,付翁(中国石油化工股份有限公司石油化工科学研究院,北京100083)
异丁烷-丁烯固体酸烷基化反应动力学研究进展
刘晓宇,周祥,郭锦标,王鑫磊,付翁
(中国石油化工股份有限公司石油化工科学研究院,北京100083)
摘要:探究了固体酸烷基化反应机理和转化规律,对其进行动力学研究具有重要的理论和现实意义。概述了固体酸烷基化反应体系具有代表性的动力学研究成果,总结了不同尺度动力学模型的建立规则和对改善催化剂失活所提供的建议,指出建立固体酸烷基化分子水平机理层面动力学模型,对本征动力学性质进行描述,反映动力学参数与原料性质、操作变量、催化剂之间存在的内在联系,可以更好地为新型催化剂研发、反应器设计和工艺流程优化提供理论支持。
关键词:烷基化;固体酸;动力学;催化剂失活;机理层面动力学模型
第一作者:刘晓宇(1984—),女,博士研究生,主要从事烷基化反应动力学研究工作。联系人:郭锦标,教授级高级工程师。E-mail guojinbiao.ripp@sinopec.com。
面对日益严重的环境污染问题,发展经济型清洁汽油燃料已是当务之急。目前我国汽油的主要成分是催化汽油和重整汽油,此外还需添加MTBE等高辛烷值组分。随着新汽油使用标准的颁发,对汽油中芳烃、烯烃、硫含量等指标的限制更为严格,亟需开发清洁的高品质汽油调合组分,要求其具有高辛烷值,又要控制对芳烃、烯烃、硫等的引入,满足环保要求。利用催化裂化产物裂化气中的异丁烷与丁烯生产的烷基化汽油,其主要成分为异辛烷,具有辛烷值高(RON95~98)、无芳烃、硫,Reid蒸汽压低等优点,是十分理想的汽油调合组分。
传统的烷基化汽油生产工艺使用的是液体酸催化剂[1],如氢氟酸、硫酸等,但是液体酸催化剂存在高酸耗、设备腐蚀、废硫酸处理困难和HF腐蚀性挥发等环境污染和安全问题。因此,国内外着力开发既可满足安全和环保要求,又易于再生的固体酸催化剂及生产工艺。目前已开展研究的固体酸催化剂主要有分子筛、杂多酸、固体超强酸[2-4]等,但由于都存在快速失活和选择性差的问题,难以实现工业化。因此更多研究关注其反应动力学,通过对反应机理进行探索,描述反应物的转化规律,可对反应器设计、工艺流程优化、新型催化材料开发提供理论支持。
1 烷基化反应机理
烷基化反应机理研究在文献中多见报道[5],遵循正碳离子反应机理,为B酸催化体系。反应中间体为正碳离子,不同于液体酸催化体系,固体酸催化体系的反应中间体为吸附态正碳离子。CORMA 等[6]利用ONIOM方法,证明在B酸中心上烯烃发生质子化反应,通过类似正碳离子结构的过渡态,然后与分子筛表面上的O原子结成烷氧键,生成相对稳定的中间体。JANIK等[7]则以杂多酸为例,应用DFT分子模拟技术,确定反应中间体正碳离子以烷氧键形式吸附在催化剂表面,并计算了上述反应的能垒,如图1所示。由此可以认为,固体酸烷基化反应中间体是与催化剂表面生成烷氧键的吸附态正碳离子。
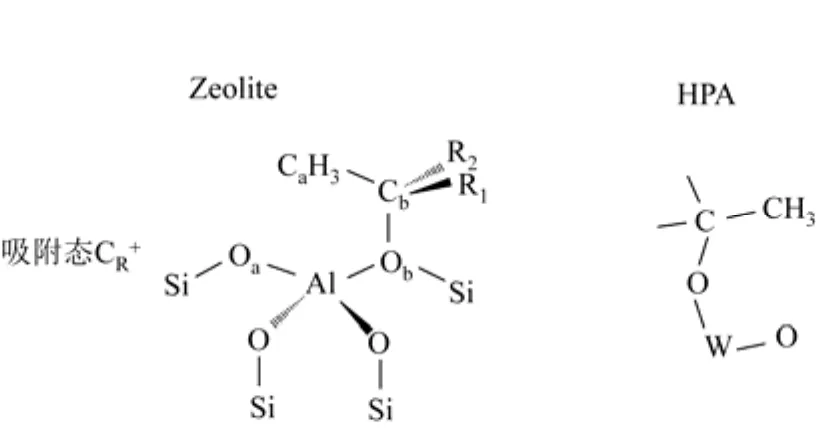
图1 固体酸催化体系中吸附态正碳离子存在形式
固体酸反应体系中包括8种基元反应类型,分别为质子化反应/去质子化反应、PCP异构反应、分子内氢/甲基转移反应、聚合反应、β-断裂反应、分子间氢转移反应(hydride transfer)。实验证明,在非高温反应条件下,烷烃不能发生脱氢和脱甲基反应,生成正碳离子[8]。
(1)质子化反应 该反应是烷基化反应的始发反应,按照Markovnikov’s规则,由烯烃双键与催化剂活性中心的氢质子发生反应,经过过渡态,生成吸附态的正碳离子中间体,反应快速达到平衡。随着催化剂酸强度增加,可以降低反应活化能,加速反应进行[9],如式(1)。

(2)去质子化反应 该反应是质子化反应的逆反应,吸附态的正碳离子发生脱附反应,生成烯烃,还原催化剂活性中心,比质子化反应具有更高的能垒,如式(2)。

(3)分子内氢转移和分子内甲基转移反应 这两类基元反应是不产生支链变化的异构化反应,发生在带有正电荷或甲基取代基的碳原子和比邻碳原子之间,如式(3)、式(4)。
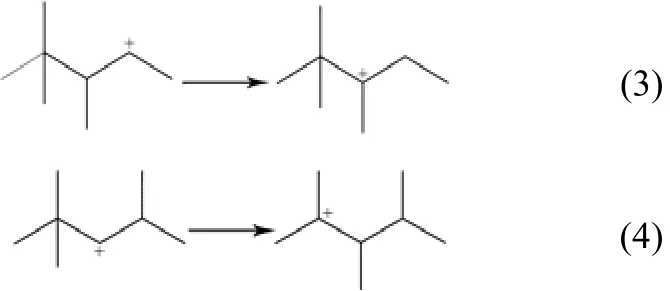
(4)PCP异构化反应 吸附态正碳离子按照Brouwer’s反应规则发生PCP异构,产生支链变化。BORONAT等[10]分别讨论直链C4+和直链C5+发生PCP反应的机理路径,发现直链C4+通过甲基环丙烷伯正碳离子过渡态,生成叔丁基正碳离子。而直链C5+则不同于直链C4+,无法经过PCP异构直接生成叔正碳离子。而是先由经过PCP异构生成仲正碳离子,而后发生分子内氢转移反应,生成叔正碳离子,如式(5)、式(6)。
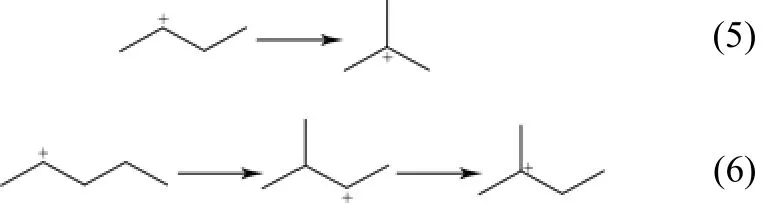
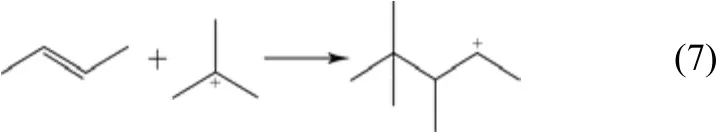
(5)聚合反应 该反应发生在正碳离子和烯烃之间,吸附态正碳离子的正电荷进攻烯烃双键,带有正电荷的碳原子与具有较少取代基的双键碳原子之间生成新的σ键,如式(7)。
(6)β-断裂反应 该反应是带有正电荷的碳原子的α位和β位之间发生断键,相对应生成小分子的烯烃和正碳离子。大分子正碳离子更易发生β-断裂反应,随着催化剂酸强度越大[10],越有利于β-断裂反应的发生,正碳离子碳链越长,越易于发生β-断裂反应,如式(8)。
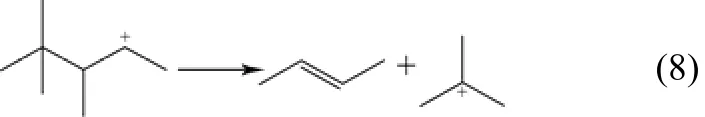
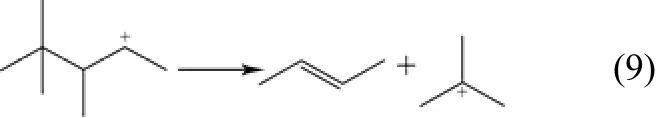
(7)分子间氢转移反应 该反应发生在B酸中心上[11],由正碳离子攻击异丁烷的C—H键,正碳离子生成相应的烷烃,异丁烷生成叔丁基正碳离子。此反应为决定催化剂失活快慢的关键步骤,如式(9)。
根据生成目标产物和副产物的反应路径,可将烷基化反应体系划分为主反应体系和副反应体系。在主反应体系中,首先由丁烯(C4=)接受B酸中心提供的氢质子,生成吸附态正碳离子(CR+)后与C4=发生聚合反应生成C8+,再与异丁烷(C40)发生分子间氢转移反应,对应生成tri-C4+和目标产物iC80。在副反应体系中,C8+可以继续与C4=发生聚合反应,生成C12+等大分子的不饱和高聚物,不易发生去质子化反应和分子间氢转移反应,导致催化剂失活。CR+还可以发生β-断裂反应,生成C3+~C9+和C3=~C9=。体系中所有CR+都可以发生分子内氢/甲基转移和PCP异构反应,并与C40发生分子间氢转移反应,所以烷基化反应体系中物种分布主要包括:C30~C90,C3=~C9=,C3+~C12+,见图2。
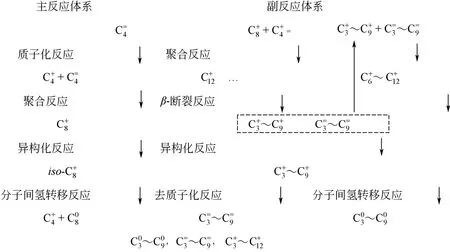
图2 烷基化反应体系物质分布
烷基化反应产物中最理想组分为三甲基戊烷(TMP),主要是由tri-C4+与丁烯聚合生成,而sec-C4+与丁烯聚合对应生成DMH(二甲基己烷),由此可见,反应中间体正碳离子的结构与目标产物分布密切相关。
引起催化剂失活的原因[12]是反应过程中生成C12+等大分子的不饱和高聚物,覆盖在催化剂活性中心上或堵塞催化剂孔道,并随着催化剂的失活,产物选择性发生变化,产物中TMP和DMH收率下降,C5~C10组分增多。
2 烷基化动力学
研究反应动力学的目的是明确化学反应速率,一是可以通过拟合反应速率与反应条件(温度、压力和组分浓度等)之间的关系,从宏观角度建立关联模型;二是实现对反应机理的验证,对本征动力学性质进行描述,从微观角度建立分子水平动力学模型,可以分为路径层面和机理层面两个层次。对于固体酸烷基化反应体系,诸多研究工作分别从宏观和微观研究反应物的转化规律进行动力学研究,建立不同尺度的动力学模型。
早期,SIMPSON等[13]从探求液-固相反应失活路径的角度,对烷基化体系进行动力学分析,建立了与反应条件关联的烯烃转化速率模型。针对主要反应:丁烯与C4+发生聚合反应(速率常数k1)、C8+发生分子间氢转移反应(速率常数k2)和丁烯与C8+发生聚合反应(速率常数k3),进行了讨论。通过实验数据,计算出烯烃与C4+聚合反应速率常数k1大于其与C8+聚合反应的速率常数k3,而k3又远远大于异丁烷与C8+发生分子间氢转移的反应速率常数k2。得出结论:由于k1>k3>k2,导致生成C12+的速率大于生成异辛烷的速率,从而引起催化剂的失活。而引起k2过小的原因是分子间氢转移反应受空间位阻影响。此外在液相反应条件下,对于超稳Y型分子筛催化剂而言,引起失活的原因还有严重的内扩散限制,并应用脉冲实验对此进行了验证,当反应物以气态参与反应,消弱内扩散影响时,可减缓失活。因此设定丁烯聚合和分子间氢转移反应为一级反应,并引入扩散系数,在不考虑催化剂失活的情况下,认为反应体系中各烃类转化近似平衡,所以将C4=的转化率与k1、k2进行关联,回归出烯烃转化率与反应条件之间的数学关系。然后以催化剂活性中心被不易脱附的高聚物(C12+)覆盖视为催化剂失活,将催化剂失活速率和异丁烷与C8+发生分子间氢转移的反应速率常数k3进行关联,以此建立的失活动力学模型。该研究从动力学的角度对固体酸失活路径进行了阐述,因此反应条件优化和反应器设计以及分子筛改性应侧重解决分子间氢转移反应的空间位阻和内扩散影响。TAYLOR等[14]进一步将反应时间作为衡量因素,建立了考虑反应时间的失活动力学模型,将总包反应IB(异丁烷)+2B(丁烯)—→Products设定为一级反应,根据阿伦尼乌斯公式求取反应速率常数。引入失活速率常数kd(kd与反应时间定量相关),以校正实际反应速率常数kw。然后将反应速率与烯烃转化率、烯烃流率和催化剂用量进行关联,求得kd和kw,进而可知对应的活化能和指前因子。该模型可用来估算不同温度下随时间变化的丁烯转化率和有效反应时间。
上述对固体酸体系的动力学研究都是在实验数据的基础上,以烯烃转化率为研究对象,回归其与反应时间、反应条件的定量关系的关联模型,并不能从反应本质出发,讨论催化剂对反应速率和方向的影响,从预测产物组成和分布的角度反映催化性能。因而NAYAK等[11]尝试将反应体系进行细致的划分,研究不同反应之间的关系,进行产物的预测。对体系中6个反应路径进行讨论,建立分子水平路径层面动力学模型,在SIMPSON等研究的基础上,对内扩散限制影响进行动力学研究。NAYAK认为体系为B酸催化,将活性中心浓度设定为B酸浓度,将生成C12+的B酸中心视为失活。应用所建模型分析可知,减小分子间氢转移反应受内扩散限制影响,可减缓催化剂失活。进而得出结论:催化剂上B酸中心以“蛋壳”(在催化剂外表面成薄层分布)形式分布可对抑制催化剂失活有积极作用,并且随着硅铝比的下降,B酸中心增加,可以增加产物中TMP的含量。增加进料烷烯比,同样可以改善催化剂失活。该模型对C8产物的预测结果与实验结果相比较,偏差在5%范围内,但该路径层面动力学模型对产物组成和收率不能实现分子水平的预测,且动力学参数依赖实验数据回归,外推性差。
SANCHEZ-CASTILLO等[15]以预测产物分子水平为目的,在机理层面对气-固相烷基化体系进行研究,建立适用于523~773K温度范围的动力学模型。认为体系中包含7种基元反应,分别为烯烃质子化、正碳离子去质子化、异构化、分子间氢转移、聚合、β-断裂、异丁烷脱氢/脱甲基反应(高温下发生)。该动力学模型将反应中间体设定为吸附态正碳离子,建立求取吸附焓的经验公式以此计算取吸附态正碳离子生成焓,其中取值为−90kJ/mol;Ha作为回归参数。根据反应机理,构建的反应网络包括277个分子和中间体,370个基元反应,共划分为4个反应族。根据反应族概念和热力学限制将动力学参数化简为12个,并利用分子模拟计算和实验数据回归的方法分别计算活化能。对于质子化反应,设定所有反应的活化能为固定值,利用分子模拟技术计算活化能为50kJ/mol;对聚合/裂化反应,根据正碳离子在反应过程中结构的变化,将活化能划分为3种,回归得到Ett=102.2kJ/mol、Est=Ess= 115.1kJ/mol;对于异构化反应,利用分子模拟技术计算活化能,其中无支链数变化异构化反应活化能为80kJ/mol,有支链数变化异构化反应活化能为110kJ/mol;对于分子间氢转移反应族,根据发生反应的正碳离子碳数分别回归活化能EC3=64.3kJ/mol、EC4=76.5kJ/mol、EC≥5=62.2kJ/mol。将计算得出的参数应用于模型,预测产物组成与实验数据趋势吻合。然后通过考察各基元反应对产物收率影响,进行灵敏度分析,将反应网络化简为31个反应,构建了涵盖主要动力学信息的反应网络。该模型对固体酸烷基化反应体系实现了机理层面的定性研究,但模型定量研究上存在偏差,没有细致讨论各基元反应反应速率的差异,体现催化剂对不同类型基元反应的催化活性。
MARTINIS等[8,16]建立的考虑催化剂失活的单事件机理层面动力学模型,对烷基化体系进行更为细致的定性研究,并着重在定量的角度,将动力学参数与催化剂性质相关联,考虑催化活性对反应速率的影响。该模型根据体系中基元反应类型共划分为7个反应族:烯烃质子化、正碳离子去质子化、分子内氢转移、分子内甲基转移、分子间氢转移、聚合、β-断裂。根据反应机理,制定反应规则,利用计算机软件生成包含3130个基元反应的反应网络。利用单事件概念、Evans-Polanyi关系式和热力学约束将3130个动力学参数化简为11个。并根据线性自由能关系,利用热力学参数计算动力学参数。因此,活化能转化为参数E0,α和反应焓变的函数,结合实验数据对参数E0、α进行回归。并通过引入稳定能,建立了计算吸附态正碳离子生成焓的新方法,将动力学参数与催化剂性质相关联。将碳数大于8的正碳离子视为积炭前体,失活函数通过活性中心被不可逆吸附正碳离子的覆盖率来表示,并在反应速率方程中引入失活系数,以此校正为实际反应速率。利用实验数据对于模型预测结果进行验证,预测结果与实验数据较好吻合。通过对各反应族活化能的计算发现,分子间氢转移活化能较大,这是造成C8+易与烯烃发生聚合反应,不易与异丁烷发生分子间氢转移反应的原因,从而引起催化剂失活,所以催化剂设计应关注具有高密度的中强酸活性中心的介孔分子筛催化剂,以改善催化剂失活。
3 结语
对于固体酸烷基化体系的动力学研究,从SIMPSON等和TAYLOR等所建立的关联模型可以看出分子筛催化剂改性应侧重解决分子间氢转移反应的空间位阻和内扩散影响。而NAYAK等所建立的路径层面动力学模型,通过对B酸催化机理验证和内扩散影响考虑,提出了对分子筛催化剂构型的改性优化和反应器设计的建设性意见,认为B酸中心以“蛋壳”(在催化剂外表面成薄层分布)形式分布可对抑制催化剂失活有积极作用,并且随着硅铝比的下降,B酸中心增加,可以增加产物中TMP的含量。SANCHEZ-CASTILLO等和MARTINIS等建立了分子水平机理层面动力学模型,更深入对本征动力学的研究,对反应网络中基元反应反应速率进行理论计算,将动力学参数与催化剂酸中心浓度和酸强度相关联,认为新型固体酸烷基化催化剂研发应关注具有高密度的中强酸活性中心的介孔分子筛催化剂,可以有效降低分子间氢转移反应活化能,改善催化剂失活。
随着研究固体酸烷基化反应体系的关注点越来越转向反应组分发生表面反应的转化规律,因此对固体酸烷基化反应体系进行分子水平机理层面动力学研究可以更好地对本征动力学性质进行描述,将动力学参数与催化剂性质相关联,以体现催化剂活性差异,反映出其与原料性质、操作变量、催化剂之间存在的内在联系,为新型固体酸催化剂研发提供定性和定量的理论支持。然而,机理层面动力学模型规模较大,存在参数计算、优化和模型求解困难等问题,部分参数仍依赖实验数据回归。因此控制模型规模合理而又不损失动力学信息和建立本征动力学参数的计算方法,是有待解决的重要问题。并且随着分析技术的发展,完善对积炭的定性分析,进一步明确失活机理,对固体酸烷基化体系的动力学研究将更为全面,在此基础上建立分子水平机理层面动力学模型将作为该领域的发展方向,可以更好地为新型催化剂研发、反应器设计和工艺流程优化提供理论支持。
参考文献
[1]WEITKAMP J,TAA Y. Isobutane/butene alkylation on solid catalysts where do we stand[J]. Catalysis Today,1999,49:193-199.
[2]CORMA A,MARTINEZ A. Chemistry,catalysts and process for isoparaffin-olefin alkylation:actual situation and future trends[J]. Catal. Rev.:Sci. Eng.,1993,35(4):83-570.
[3]何奕工,舒兴田. 超临界流体状态下的异构烷烃与烯烃烷基化反应[J]. 催化学报,1999,40(3):403-408.
[4]CORMA A,JUAN-RAJADELL M I,LOPEZ-NIETO J M,et al. A comparative study of O42−/ZrO2and zeolite beta as catalysts for the isomerization of n-butane and the alkylation of isobutane with 2-butene[J]. Applied Catalysis A,1994,111(2):175-189.
[5]FELLER A,ZUAZO I,GUZMAN A. Common mechanistic aspects of liquid and solid acid catalyzed alkylation of isobutane with n-butene[J]. Journal of Catalysis,2003,216:313-323.
[6]BORONAT M,VIRUELA P M,CORMA A. Reaction intermediates in acid catalysis by zeolites:prediction of the relative tendency to form alkoxides or carbocations as a function of hydrocarbon nature and active site structure[J]. J. Am. Chem. Soc.,2004,126:3300-3309.
[7]JANIK M J,DAVIS R J,NEUROCK M. A density functional theory study of the alkylation of isobutane with butene over phosphotungstic acid[J]. Journal of Catalysis,2006,224:65-77.
[8]MARTINIS J M,FROMENT G F. Alkylation on solid acids.Part 1.Experimental investigation of catalyst deactivation[J]. Ind. Eng. Chem. Res.,2006,45:940-953.
[9]FANG H,ZHENG A,LI S,et al. New insights into the effects of acid strength on the solid acid catalyzed reaction:theoretical calculation study of olefinic hydrocarbon protonation reaction[J]. J. Phys. Chem. C,2010,114(22):10254-10264.
[10]BORONAT M,VIRUELA P,CORMA A. Theoretical study of the mechanism of branching,rearrangement of carbenium ions[J]. Applied Catalysis A:General,1996,146:207-223.
[11]NAYAK S V,RAMACHANDRAN P A,DUDUKOVIC M P. Modeling of key reaction pathways:zeolite catalyzed alkylation processes[J]. Chemical Engineering Science,2010,65:335-342.
[12]BARTHOLOMEW C H. Mechanisms of catalyst deactivation[J]. Applied Catalysis A:General,2001,212:17-60.
[13]SIMPSON M F,WEI J,SUNDARESAN S. Kinetic analysis of isobutane/butene alkylation over ultrastable H-Y zeolite[J]. Ind. Eng. Chem. Res.,1996,35:3861-3873.
[14]TAYLOR R J,SHERWOOD JR D E. Effects of process parameters on isobutane/2-butene alkylation using a solid acid catalyst[J]. Applied Catalysis A:General,1997,155:195-215.
[15]SANCHEZ-CASTILLO M A,AGARWAL N,MILLERET C,et al. Reaction kinetics study and analysis of reaction schemes for isobutane conversion over USY zeolite[J]. Journal of Catalysis,2002,205:67-85.
[16]MARTINIS J M,FROMENT G F. Alkylation on solid acids. Part 2.Single-event kinetic modeling[J]. Ind. Eng. Chem. Res.,2006,45:954-967.
研究开发
Advances in the research development of isobutane/butene solid acid-catalyzed alkylation kinetics
LIU Xiaoyu,ZHOU Xiang,GUO Jinbiao,WANG Xinlei,FU Weng
(Research Institute of Petroleum Processing,SINOPEC,Beijing 100083,China)
Abstract:Kinetic analysis for understanding the reaction mechanism and hydrocarbon transformations rules has both theoretical and practical significance. The typical development and main research methods at different scales for alkylation kinetics are reviewed in this paper. Recommendations for improving the catalyst are made. The molecular-based kinetic modeling could provide detailed and explicit descriptions of the intrinsic kinetic properties,which reflect the relationship between the kinetic parameters and the feedstock,reaction conditions and catalyst. Mechanistic kinetic modeling could show important theoretical support for the optimization of reactor configuration,technological process and catalysts research.
Key words:alkylation; solid acid; kinetics; catalyst deactivation; mechanistic kinetic modeling
中图分类号:TE 624.4
文献标志码:A
文章编号:1000–6613(2016)04–1007–05
DOI:10.16085/j.issn.1000-6613.2016.04.006
收稿日期:2015-08-12;修改稿日期:2015-11-01。
