Al12X(X=Cu,Ag,Au)团簇的稳定结构和磁性
2016-05-05刘莲莲李宝兴
金 鑫,刘莲莲,李宝兴
(杭州师范大学理学院,浙江 杭州 310036)
Al12X(X=Cu,Ag,Au)团簇的稳定结构和磁性
金 鑫,刘莲莲,李宝兴
(杭州师范大学理学院,浙江 杭州 310036)
本文用基于第一性原理的阿姆斯特丹密度泛函程序系统地研究了Al12X(X=Cu,Ag,Au)团簇的稳定结构和磁性.研究表明Cu原子位于截角20面体的中心位置,而Ag和Au原子则占据表面位置.3个团簇的磁矩均为3μB,它们取决于电子结构.图示发现3个团簇的最高占据轨道的电子云分布图相似,但在最高占据轨道以下的电子云分布图中,Al12Ag和Al12Au团簇的非常相似.
Al13团簇;替换;稳定性;基态结构
0 引 言
团簇是介于原子与大块宏观物质之间的一种特殊物态形式,其物理和化学性质既不同于原子又不同于块状物体.它们最鲜明的特征是其物化性质随原子数而变化,所出现的许多奇异特性又展示出巨大潜在的应用价值.因此,最近几十年来,科学家们付出了巨大努力,采用各种手段和方法研究它们的物化性质.

尽管对Al13团簇的掺杂研究已有一些报道,但到目前为止这些研究还非常有限,特别是对掺贵金属Ag和Au原子的磁性研究报道几乎没有,就是对它们的结构研究结果也不尽相同,甚至互相矛盾.为此,很有必要用其它方法对Al12M(M=Cu, Ag, Au)团簇的稳定结构进行研究,尤其是对它们的磁性进行研究.据笔者所知,除了在Al13团簇中掺铜的磁性有过一些报道外,但未见掺银和金原子的磁性报道.
本文采用基于第一性原理的阿姆斯丹特密度泛函程序,研究用一个Cu、Ag和Au原子在Al13团簇中进行替换掺杂所得结构的稳定性和磁性.
1 计算方法
阿姆斯特丹密度泛函程序(Amsterdam Density Functional, 简称ADF)采用了基于第一性原理的密度泛函理论[9], 是目前国际上公认的用于团簇等研究的一种先进商业计算程序.在计算过程中, 考虑了广义梯度近似(GGA),其中采用了Becke-Perdew(B-P)交换关联泛函.对Al和Cu等金属原子所计算的Hartree-Fock交换能与它们的精确值非常吻合,其均方根偏差仅仅为0.11%[10].
用自编程序随机产生了上万个Al13团簇的几何结构,用ADF程序对这些结构进行结构优化,一些结构不收敛,还有一些有多个结构收敛于同一个结构.进一步对具有较大结合能的几个结构提高精度再次进行结构优化,并进行了频谱计算,消除虚频.最后,分析比较得到了Al13团簇的基态结构.在此基础上在Al原子位置上用Cu、Ag和Au原子替换其中一个Al原子,并用上述程序对该初始构型进行结构优化,得到了各种稳定的Al12X掺杂结构.另外,也尝试了在Al12团簇上吸附一个杂质原子,或用12个Al原子和1个Cu、Ag或Au原子随机产生初始结构并进行结构优化,最后得到的基态结构是相同的.用ADF程序我们已经研究了一些团簇的吸附或掺杂性质[7,11-12],发现了一些有趣的现象.
2 结果与讨论
Al13团簇存在大量的稳定结构,我们曾经讨论过6个代表性的稳定结构[7],目前国际上公认的Al13团簇的基态结构是一个截角20面体,它畸变前具有Ih对称性.中性的Al13团簇结构稍有结构畸变,对称性有所降低,但它的负离子结构,有40个价电子,非常稳定,是一个完美的截角20面体结构,具有Ih对称性.Al13团簇的基态结构如图1所示.
作为对所用方法的一种检验,表1给出了用不同方法获得的Al13团簇表面上原子之间的键长(Å)、计算的团簇结合能和实验值.这些结果表明所用的方法适合于铝团簇以及掺杂铝团簇的研究.
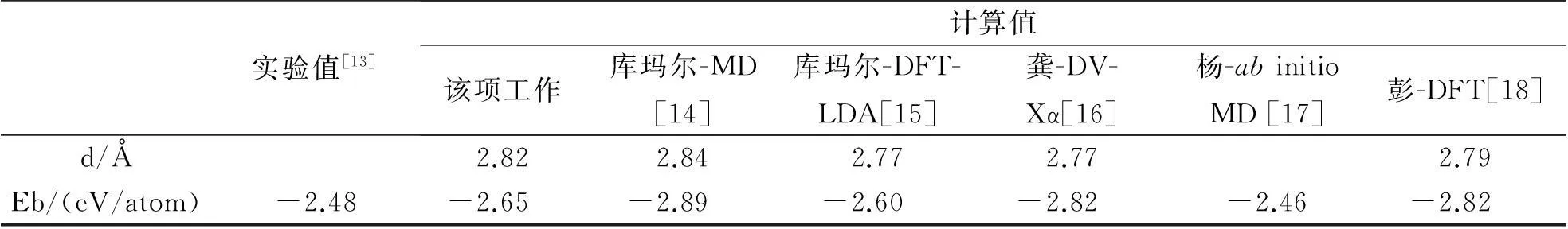
表1 不同方法获得的Al13团簇表面原子之间的键长(Å)、计算的结合能和实验值
Al13团簇的结合能为34.44eV.布居分析表明表面原子上有11.27个电子的电荷量转移到中心铝原子上,即平均每个表面原子贡献0.94个电子的电量.尽管Al13团簇是电中性的,但由于表面原子带正电,这使得该团簇的表面容易吸引电子.当它获得一个电子成为负离子后,中心原子的电荷量从 11.27e 升到11.89e,这意味着被吸引的电子有60%的电量转移到中心原子上.较大的库伦引力使得这个截角20面体结构更致密.显然,该结构有2个不同的位置,一个是表面上几乎等价的12个原子的位置,另一个是中心原子的位置.用铜、银和金3种不同原子分别替代中心原子和表面原子,然后进行结构优化.结构优化后得到的最稳定结构如图2所示,左、中和右分别对应于铜、银和金原子的掺杂结构图.
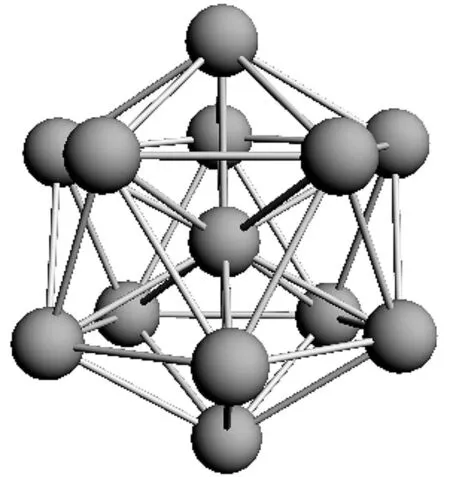
图1 Al13团簇的基态结构Fig. 1 The ground state structureof Al13 cluster

图2 Al12Cu、Al12Ag和Al12Au团簇的基态结构Fig. 2 The ground state structures of Al12Cu、Al12Ag and Al12Au clusters
掺杂铝团簇Al12Cu、Al12Ag和Al12Au的结合能和磁矩如表2所示.从图2和表2可以看出,掺铜、银和金原子后所获得的最稳定结构与纯铝Al13团簇的结构形似,但它们的结合能依次降低,而磁矩都为3μB.3个结构中,Al12Cu团簇的对称性最高,为Ih.而Al12Ag和Al12Au团簇的对称性均为Cs.铜原子处在中心位置时最稳定,而银和金原子则位于表面位置上最稳定.这与原子的大小有关,铜原子的半径比铝原子的半径要稍小,它位于中心会增加健能,使表面铝原子结合得更为紧密.Al12Cu团簇也是非常完美的截角20面体结构.银和金原子的原子半径比铝原子的半径大约要大10%,这使得它们位于表面位置处更为稳定.
表2还给出了这3个团簇中铜、银和金原子的局域磁矩ML,最高占据轨道(HOMO)与最低未被占据轨道(LUMO)之间的能隙Eg,杂质原子所带电量Qi,中心原子所带电量Qc,从这些数据可以看出:掺银和金原子所得的结果相近.图3给出了Al12Cu、Al12Ag和Al12Au团簇中几个最高占据轨道的电子云图.显然,3个团簇的最高占据轨道的电子云图非常相似,但次最高占据轨道的电子云图就有差别,Al12Ag和Al12Au团簇的非常相近,它们与Al12Cu团簇的不同.为便于比较,我们优化了铜原子处于表面位置上的几何结构,该结构中磁矩为3μB的态不是基态,但它最高占据轨道的电子云图与图3中的非常相似,这表明磁矩为3μB的截角20面体结构的最高占据轨道的电子云图主要取决于它的几何结构,而与该结构中掺杂原子的位置没有太大关系.研究表明,在总原子数小于14的这些掺杂团簇中,只有13个原子的截角20面体结构表现出磁性,而同样是13个原子的其他几何结构也没有出现磁性,这表明掺杂团簇的磁性与这个特定的几何结构有关.在这个特定的结构中,贵金属原子不一定要处在中心位置上.铜原子处在中心位置上最稳定,而银和金原子则在表面位置上最稳定.

表2 Al12Cu、Al12Ag和Al12Au团簇的结合能、磁矩和能隙等
在参考文献[19]中,Kumar等人报道了Al12Cu团簇高磁矩与洪特定则有关.除去铜原子上10个3 d电子,Al12Cu团簇中有37个价电子,填满1f壳层需要34个电子,根据洪特定则,剩下的3个电子将填在凝胶模型中3个等价的2p态上,它们平行排列,形成磁矩3μB.掺银和金原子,虽然它们处在表面位置上,但磁矩为3μB的态仍是最稳定的.可见,这些结构的稳定性和磁矩主要取决于它们的电子结构.


图3 Al12Cu、Al12Ag和Al12Au团簇几个最高占据轨道的电子云图Fig. 3 The electronic clouds of several HOMOs in Al12Cu、Al12Ag and Al12Au clusters
3 结 论
本文用阿姆斯特丹密度泛函程序系统地研究了Al12X(X=Cu,Ag,Au)团簇的稳定结构和磁性.研究表明,铜原子处在团簇的中心位置上,而银和金原子则位于表面位置上,但它们的磁矩均为3μB.Al12Cu,Al12Ag和Al12Au团簇的结合能依次减小,掺Cu原子的结构对称性为Ih,而掺Ag和Au原子的结构对称性均为Cs.三个团簇的磁性取决于团簇的电子结构.
[1] 彭平,李贵发,郑采星,等.Aln(n=3,4,6,13,19)团簇的结构稳定性与形态演化[J].中国科学E辑(技术科学),2006,36(9):975-982.
[2] RAO B K, JENA P. Evolution of the electronic structure and properties of neutral and charged aluminum clusters: A comprehensive analysis[J]. J Chem Phys,1999,111(5):1890-1904.
[3] LI X, WU H, WANG X B, et al. S-p Hybridization and Electron Shell Structures in Aluminum Clusters: A Photoelectron Spectroscopy Study[J]. Phys Rev Lett,1998,81(9):1909.
[4] PAL R, LI F C, BULUSU S, et al. Probing the electronic and structural properties of doped aluminum clusters: MAl12(M=Li, Cu, and Au)[J]. J Chem Phys,2008,128(2):024305.
[5] LI X, WANG L S. Experimental search and characterization of icosahedral clusters: Al12X-(X=C,Ge,Sn,Pb)[J]. Phys Rev B,2002,65(15):153-404.
[6] 阿巴拜克里·吐尔孙,姜园园,文静,等.“密度泛函理论研究混合团簇Al12X(X=Sc、Ti、V、Gr、Mn、Fe、Co、Ni、Cu、Zn)的结构和磁性[J].原子与分子物理学报,2013,30(2):216-222.
[7] 陆霞,孙颖,李宝兴,等.外来吸附原子对Al13团簇的结构影响[J].杭州师范大学学报(自然科学版),2014,13(5):556-560.
[8] ZOPE R R, BARUAH T. Conformers of Al13, Al12M, and Al13M (M=Cu, Ag, and Au) clusters and their energetics[J]. Phys Rev A, 2001,64(5):053202.
[9] VAN LENTHE E, BAERENDE E J. Optimized Slater-type basis sets for the elements 1-118[J]. J Comput Chem, 2003,24(9):1142-1156.
[10] BECKE A D. Density-fnnctional exchange-energy approximation with correct asymptotic behavior[J]. Physical Review A, 1988,38(6):3098-3100.
[11] 顾娇娇,李宝兴,马志伟.氮掺杂C20富勒烯的几何结构和稳定性[J]. 杭州师范大学学报(自然科学版),2013,12(2):140-144.
[12] 马志伟,李宝兴,顾娇娇. CmNn(m,n=1-10,4≤m+n≤11)团簇的结构和稳定性研究[J]. 杭州师范大学学报(自然科学版),2013,12(4):359-364.
[13] RAY U, JARROLD M F, BOWER J E, et al. Photodissociation kinetics of aluminum cluster ions: Determination of cluster dissociation energies[J]. J Chem Phys,1989,91(5):2912-2921.
[14] KUMAR V. Structure and electronic properties of Al14and Al13Na clusters[J]. Phys Rev B,1998,57(15):8827-8829.
[15] KUMAR V, BHATTACHARJEE S, KAWAZOE Y. Silicon-doped icosahedral, cuboctahedral, and decahedral clusters of aluminum[J]. Phys Rev B,2000,61(12):8541.
[16] GOGN X G, KUMAR V. Enhanced stability of magic clusters: A case study of icosahedral Al12X, X=B, Al, Ga, C, Si, Ge, Ti, As[J]. Phys Rev Lett,1993,70(14):2078-2081.
[17] YNAG S Y, DRABOLD D A, ADAMS D A, et al. First-principles local-orbital density-functional study of Al clusters[J]. Phys Rev B,1993,47(3):1567-1576.
[18] PENG P, LI G F, ZHENG C X, et al. Structure stability and configuration evolution of Aln(n=3, 4, 6, 13, 19) clusters[J].Science in China Series E (Technological Sciences),2006,49(4):385-392.
[19] KUMAR V, KAWAZOE Y. Hund’s rule in metal clusters: Prediction of high magnetic moment state of Al12Cu from first-principles calculations[J]. Phys Rev B,2001,64(11):115405.
The Stable Structure and Magnetism of Al12X(X=Cu,Ag,Au)Clusters
JIN Xin, LIU Lianlian, LI Baoxing
(School of Science, Hangzhou Normal University, Hangzhou 310036, China)
In this paper, the stable structures and magnetism of Al12X(X=Cu, Ag, Au)clusters are studied by Amsterdam Density Functional based on the First-principles. The results show that Cu atom lies at the centre of the icosahedral structure, whereas Ag and Au atoms occupy the surface. The three clusters have a average magnetic moment of 3μB, which is determined by their electronic structures. It is found that the electron cloud distribution maps on the highest occupied molecular orbital of the three clusters are similar, but the electron cloud distribution maps of Al12Ag and Al12Au clusters are very similar below the highest occupied molecular orbital.
Al13cluster; substitution; stability; ground state structure
2015-06-02
教育部“优博”基金项目(200320);浙江省自然科学基金项目(Y6100098).
李宝兴(1960—),男,教授,博士,主要从事团簇研究.E-mail: phybxli@hznu.cdu.cn
10.3969/j.issn.1674-232X.2016.02.013
O469
A
1674-232X(2016)02-0192-05
