沉淀方式对沉淀法制备Co3O4催化N2O直接分解性能的影响
2016-04-25胡晓波张翘楚桂佳会崔雨萌王永钊赵永祥
胡晓波,石 晶,张翘楚,桂佳会,崔雨萌,王永钊,赵永祥
(山西大学化学化工学院 精细化学品教育部工程研究中心,山西 太原030006)
沉淀方式对沉淀法制备Co3O4催化N2O直接分解性能的影响
胡晓波,石晶,张翘楚,桂佳会,崔雨萌,王永钊*,赵永祥*
(山西大学化学化工学院 精细化学品教育部工程研究中心,山西 太原030006)
摘要:以Co(NO3)2·6H2O为钴源,K2CO3为沉淀剂,采用沉淀法制备Co3O4催化剂,用于催化N2O直接分解反应。利用N2-物理吸附、XRD、FT-IR、TEM、TPR和ICP等对其进行表征,考察沉淀方式对Co3O4催化剂结构及其催化性能的影响。结果表明,沉淀方式对制备的Co3O4催化剂织构性质、物相组成和晶粒尺寸等影响不大,但显著影响其K残留量和还原性能,进而决定催化剂直接催化分解N2O的催化性能。反加法制得的催化剂中K残留量为1.43%,明显高于正加法,同时催化剂中Co(3+)较正加法更易还原,因而表现出更高的催化性能。在空速10 000 h(-1)和N2O体积分数0.1%的条件下,反加法制备的催化剂可在280 ℃催化N2O完全分解,较正加法低20 ℃。
关键词:催化剂工程;Co3O4;沉淀方式;反加法;正加法;N2O催化分解
CLC number:O633.36;TQ426.6Document code: AArticle ID: 1008-1143(2016)01-0041-06
N2O曾长期被认为是一种对环境无害的气体,并广泛应用于医学及工业等领域。近年来,随着对N2O认识和研究的不断深入,其环境危害性已得到共识。N2O不仅可破坏臭氧层[1],同时还是一种温室气体,其全球升温潜能值分别是CO2的310倍和CH4的21倍[2]。2005年2月16日开始执行的《京都议定书》中提出,限制CO2、CH4、N2O、O3、氢氟氯碳化物类和全氟碳化物等6种重要温室气体的排放[3]。因此,N2O的排放控制和去除已成为各国必须面对的一个重要课题, 其相关技术的研究和开发受到关注。
N2O主要来源于农业土壤开采,工业己二酸、硝酸及化肥的生产[4-5],使用硝酸为氧化剂的化工过程、流化床中煤的燃烧[6]以及汽车尾气NOx的消除过程[7]等。N2O催化消除的方法主要有直接催化分解法和选择性催化还原法。直接催化分解法是一种比较经济有效的方法,对催化体系的研究集中在金属催化剂[8-9]、分子筛催化剂[10-11]和氧化物催化剂[12-13],其中含钴的金属氧化物催化剂因制备方法简单、组成易于调变且具有较高的催化活性而受到关注[14-17]。
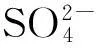
1实验部分
1.1试剂
Co(NO3)2·6H2O,分析纯,天津市大茂化学试剂厂;K2CO3,分析纯,天津市凯通化学试剂有限公司。
1.2催化剂制备
正加法:称取一定量Co(NO3)2·6H2O溶于蒸馏水中配制溶液,室温下将2 mol·L-1的K2CO3溶液匀速加入Co(NO3)2溶液中,同时剧烈搅拌,直至体系pH=9,继续搅拌2 h,静置老化2 h,120 ℃干燥12 h,将干燥的样品在400 ℃焙烧2 h制得催化剂样品,标记为Co-NP。
反加法:将Co(NO3)2溶液加至2 mol·L-1的K2CO3溶液中,调整体系pH=9,其余步骤相同,制得的催化剂标记为Co-RP。
催化剂经压片筛分,取粒径(40~60)目用于活性评价。
1.3催化剂表征
低温N2-物理吸附测定在美国麦克仪器公司ASAP-2020型物理吸附仪上进行,催化剂预先在150 ℃真空条件下脱气预处理5 h,然后在液氮浴条件下进行N2吸附-脱附测定,分别用BET公式和BJH模型计算比表面积和孔径分布。
采用德国布鲁克公司D8-Advance型X射线粉末衍射仪进行物相分析,CuKα,Ni滤波,工作电压40 kV,工作电流40 mA,2θ=10°~80°,步幅0.02°,λ=0.15 418 nm,扫描速率2.4 °·min-1,万特探测器检测。
采用德国布鲁克公司Tensor27傅里叶变换红外光谱仪进行红外表征,样品与溴化钾以质量比1∶100混合,研磨制片,室温下对催化剂进行测定,分辨率4 cm-1,扫描范围(400~4 000) cm-1。
采用美国麦克仪器公司AutochemⅡ2920型化学吸附分析仪进行TPR表征,将(40~60)目催化剂30 mg置于反应管,通入5%H2-N2混合气,流量20 mL·min-1,待基线平稳后,以10 ℃·min-1的升温速率程序升温至700 ℃,采用热导池检测器(TCD)检测耗氢量。
采用日本Jeol公司JEM-2100型透射电子显微镜观察催化剂的形貌、粒径大小以及晶面暴露,测试前,取粉末状样品先用乙醇分散,经超声波超声30 min使其分散均匀,然后用滴管移取2滴,滴定在铜网格中的碳支持膜上,最后进行分析,加压电压200 kV。
采用美国Thermo公司IRIS 1000型电感耦合等离子体光谱仪进行K含量的测定,将一定量的新鲜催化剂在加热条件下边搅拌边加浓硝酸和少量浓盐酸,直至催化剂全部溶解,定容于50 mL容量瓶中进行分析测定。
1.4催化剂活性评价
N2O催化分解活性评价在连续流动微反装置上进行,催化剂用量0.3 g,0.1%N2O,Ar为平衡气,气体总流量50 mL·min-1,空速10 000 h-1,反应物和产物采用装有Propark Q填充柱的GC-930型气相色谱仪检测,TCD检测器,载气为He,催化活性用N2O转化率评价。
2结果与讨论
2.1催化活性评价结果
不同沉淀方式制备的Co3O4催化剂催化N2O直接分解活性如图1所示。由图1可以看出,两种催化剂上N2O转化率均随反应温度升高呈明显上升趋势,但在相同反应温度时,反加法制备的催化剂上N2O转化率高于正加法,160 ℃时催化剂Co-RP上N2O即开始转化,反应温度升至240 ℃时N2O转化率已达50%,而正加法制备的催化剂Co-NP上直到200 ℃时N2O才出现极少量转化,达到50%转化率则需要270 ℃,较催化剂Co-RP高30 ℃。就两种催化剂上N2O的完全转化温度而言,催化剂Co-RP上仅为280 ℃,而催化剂Co-NP上则高达300 ℃。反加法制备的Co-RP催化剂N2O催化分解活性明显优于正加法,表明沉淀方式对沉淀法制备的Co3O4催化剂N2O直接催化分解活性有显著影响。
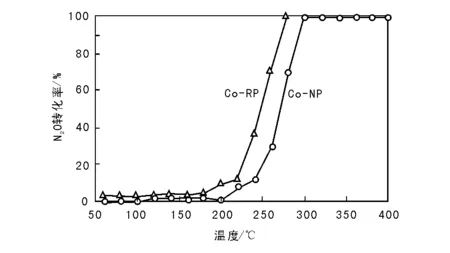
图 1 不同沉淀方式制备的Co3O4催化剂催化N2O直接分解性能Figure 1 Influence of different precipitation ways on catalytic performance of Co3O4 catalysts for direct decomposition of N2O
2.2N2-物理吸附与ICP分析
图2为采用不同沉淀方式制备的Co3O4催化剂的N2-物理吸附曲线,表1为催化剂比表面积、孔结构和ICP数据。
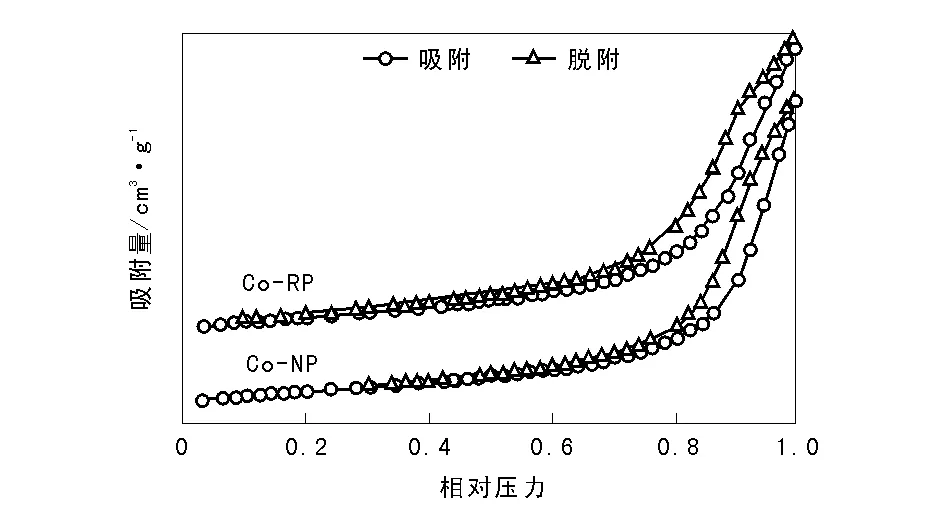
图 2 不同沉淀方式制备的Co3O4催化剂N2-物理吸附等温曲线Figure 2 N2-physisorption isotherms of Co3O4 catalysts prepared by different precipitation ways

催化剂比表面积/m2·g-1平均孔径/nm孔容/mL·g-1K残留量/%Co-RP58170.251.43Co-NP65140.241.25
从图2可以看出,两种催化剂的吸附-脱附曲线兼有Ⅲ型和Ⅴ型等温线的特征,均出现回滞环,表现出部分介孔结构特征,其主要由催化剂颗粒堆积而成。由表1可知,与正加法制得的催化剂Co-NP相比,反加法制得的催化剂Co-RP平均孔径略有增大,孔容相差无几,但比表面积略有降低,表明沉淀方式对沉淀法制备的Co3O4催化剂织构性质影响不大。与二者催化N2O直接分解性能相关联,可认为Co3O4的织构性质并不是造成两种催化剂活性差异的主要因素。刘畅等[16]在研究碱土金属对钴铈复合氧化物催化N2O分解性能影响时也发现,添加不同碱土金属后催化剂的比表面积大小顺序与其催化活性顺序不一致,认为催化剂比表面积不是影响催化活性的主要因素。两种催化剂的ICP分析结果表明,反加法制备的催化剂Co-RP中K残留量为1.43%,正加法制备的催化剂Co-NP中K残留量略低,为1.25%。
2.3XRD
图3为采用不同沉淀方式制备的Co3O4催化剂的XRD图。

图 3 不同沉淀方式制备的Co3O4催化剂的XRD图Figure 3 XRD patterns of Co3O4 catalysts prepared by different precipitation ways
由图3可以看出,Co-RP和Co-NP催化剂均在2θ=18.9°、31.2°、36.7°、44.7°、59.5°和69.7°处出现了明显的衍射峰,为尖晶石相Co3O4的特征衍射峰(JCPDS 43-1003),分别对应于Co3O4的(111)、(220)、(311)、(400)、(511)和(440)晶面。根据XRD图中(311)晶面衍射峰,利用谢乐公式计算出两种催化剂的晶粒尺寸,正加法制备的催化剂Co-NP为19.5 nm;反加法制备的催化剂Co-RP略大,为21.7 nm。可见不同沉淀方式对制备的Co3O4催化剂物相结构无明显影响,但正加法制备的催化剂晶粒尺寸略小。
2.4IR
图4为采用不同沉淀方式制备的Co3O4催化剂的红外谱图。
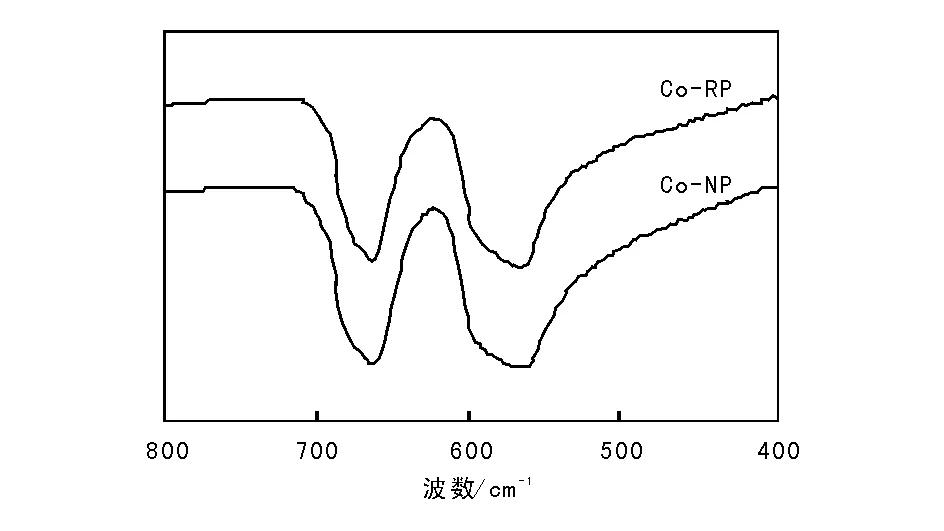
图 4 不同沉淀方式制备的Co3O4催化剂FT-IR谱图Figure 4 FT-IR spectra of Co3O4 catalysts prepared by different precipitation ways
由图4可以看出,Co-RP和Co-NP催化剂在567 cm-1和663 cm-1处出现明显的吸收峰,是由钴离子-氧键伸缩振动引起,归属为尖晶石结构的特征吸收峰[21]。567 cm-1处吸收峰归因于Co3O4结构中处于八面体位的Co3+引起的Co3+-O 振动,663 cm-1处吸收峰是由处于四面体位的Co2+的Co2+-O振动引起。对比两种催化剂的吸收峰位置、峰型及峰强度均无明显变化,表明正加法和反加法制备的催化剂具有相同的尖晶石结构,与XRD结果一致。
2.5TPR
图5为不同沉淀方式制备的Co3O4催化剂的TPR曲线。
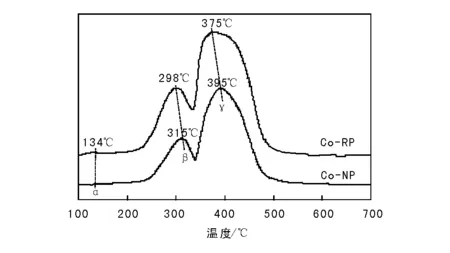
图 5 不同沉淀方式制备的Co3O4催化剂的TPR曲线Figure 5 TPR profiles of Co3O4 catalysts prepared by different precipitation ways
由图5可以看出,反加法制备的Co-RP催化剂在134 ℃、298 ℃和375 ℃出现3个还原峰,分别归属为表面氧的还原峰α,Co3+到Co2+的还原峰β和Co2+到Co0的还原峰γ[22]。正加法制备的Co-NP催化剂则在315 ℃和395 ℃出现两个还原峰,前者归属于Co3+还原为Co2+的还原峰β,后者归属于Co2+还原为Co0的还原峰γ。
与Co-NP催化剂相比,Co-RP催化剂的两个还原峰β和γ均明显向低温方向移动。Asano K等[15]研究发现,K与Co3O4间的相互作用可促进Co3+到Co2+的还原。结合本文催化剂的ICP表征结果可知,Co-RP催化剂中K残留量高于Co-NP催化剂,可以推测,反加法制备的催化剂中相对较高含量的K进一步促进了催化剂中Co3+的还原。结合N2O的分解机理[23]可知,Co3+到Co2+的还原过程是N2O直接催化分解的关键步骤,亦即Co2+为反应活性中心。Co3+越易还原为Co2+,则越有利于在催化剂中形成Co2+活性位,进而提高N2O直接分解催化活性。
结合TPR和ICP表征结果、N2O分解机理以及相关文献报道可知,反加法制备的Co3O4催化剂中残留有更高含量的K,其与Co3O4间的作用明显促进了Co3+到Co2+的还原,显著提高了Co3O4的N2O直接分解活性。
2.6TEM与HRTEM
图6为不同沉淀方式制备的Co3O4催化剂的TEM和HRTEM照片。由图6的TEM照片可以看出,反加法制得的Co-RP催化剂晶粒略有聚集,且晶粒尺寸相对较大,约(20~23) nm,而正加法制得的Co-NP催化剂分散度略好,晶粒尺寸约19 nm,与XRD结果一致。这是由于正加法制备催化剂过程中将沉淀剂滴入盐溶液,随溶液pH升高,逐步形成沉淀,同时伴随剧烈搅拌,促使沉淀前驱物较好的分散,从而使最终Co3O4催化剂分散良好;而反加法过程中将盐溶液快速加入到沉淀剂溶液中,沉淀物晶核短时间内快速形成并生长,造成最终催化剂一定程度上的轻微聚集。由图6的HRTEM照片可以看出,两种催化剂均暴露了晶面间距为0.24 nm、0.28 nm和0.46 nm的晶面,分别对应于Co3O4的(311)、(220)和(111)晶面,表明不同沉淀方式制备的Co3O4催化剂粒径形貌、分散度以及晶面暴露等区别不大。

图 6 不同沉淀方式制备的Co3O4催化剂TEM和HRTEM照片Figure 6 TEM and HRTEM images of Co3O4 catalysts prepared by different precipitation ways
3结论
(1) 沉淀方式对Co3O4催化剂的织构性质、物相组成以及粒径形貌等影响不大,但对催化剂上K残留量有一定的影响,进而使催化剂的还原性能产生较大区别,并最终决定了催化剂的N2O直接分解催化性能。
(2) 反加法制得的催化剂Co-RP中K残留量高于正加法制得的催化剂Co-NP,虽然比表面积略低,但还原性能更高,因而在催化N2O直接分解反应中表现出优异的催化性能。
参考文献:
[1]Dickinson R E,Cicerone R J.Future global warming from atmospheric trace gases[J].Nature,1986,319:109-115.
[2]联合国环境规划署.世界环境数据手册[M].北京:中国科学技术出版社,1990:19-20.
[3]吴晓蔚,朱法华,杨金田,等.火力发电行业温室气体排放因子测算[J].环境科学研究,2010,23(2):170-176.
Wu Xiaowei,Zhu Fahua,Yang Jintian,et al.Measurements of emission factors of greenhouse gas(CO2,N2O) from thermal power plants in China[J].Research of Environmental Sciences,2010,23(2):170-176.
[4]Pérez-Ramírez J,Kapteijn F,Schöffel K,et al.Formation and control of N2O in nitric acid production[J].Applied Catalysis B:Environmental,2003,44(2):117-151.
[5]Pérez-Ramírez J.Prospects of N2O emission regulations in the European fertilizer industry[J].Applied Catalysis B:Environmental,2007,70(1):31-35.
[6]Galle M,Agar D W,Watzenberger O.Thermal N2O decomposition in regenerative heat exchanger reactors[J].Chemical Engineering Science,2001,56(4):1587-1595.
[7]Becker K H,Lörzer J C,Kurtenbach R,et al.Nitrous oxide(N2O) emissions from vehicles[J].Environmental Science & Technology,1999,33(22):4134-4139.
[8]Dacquin J P,Dujardin C,Granger P.Surface reconstruction of supported Pd on LaCoO3:consequences on the catalytic properties in the decomposition of N2O[J].Journal of Catalysis,2008,253(1):37-49.
[9]Doi K,Wu Y Y,Takeda R,et al.Catalytic decomposition of N2O in medical operating rooms over Rh/Al2O3,Pd/Al2O3,and Pt/Al2O3[J].Applied Catalysis B:Environmental,200l,35(1):43-51.
[10]Pirngruber G D,Roy P K,Prins R.The role of autoreduction and of oxygen mobility in N2O decomposition over Fe-ZSM-5[J].Journal of Catalysis,2007,246(1):147-157.
[11]Smeets P J,Meng Q,Corthals S,et al.Co-ZSM-5 catalysts in the decomposition of N2O and the SCR of NO with CH4:influence of preparation method and cobalt loading[J].Applied Catalysis B:Environmental,2008,84(3/4):505-513.
[12]Giecko G,Borowiecki T,Gac W,et al.Fe2O3/Al2O3catalysts for the N2O decomposition in the nitric acid industry[J].Catalysis Today,2008,137(2):403-409.
[13]Cimino A,Indovina V.Activity of Mn3+and Mn4+ions dispersed in MgO for N2O decomposition[J].Journal of Catalysis,1973,17(1):54-70.
[14]Yoshino H,Ohnishi C H,Hosokawa S,et al.Optimized synthesis method for K/Co3O4catalyst towards direct decomposition of N2O[J].Journal of Materials Science,2011,46(3):797-805.
[15]Asano K,Ohnishi C,Iwamoto S,et al.Potassium-doped Co3O4catalyst for direct decomposition of N2O[J].Applied Catalysis B:Environmental,2008,78(3):242-249.
[16]刘畅,薛莉,贺泓.碱土金属对钴铈复合氧化物催化剂催化N2O分解的影响[J].物理化学学报,2009,25(6):1033-1039.
Liu Chang,Xue Li,He Hong.Influence of alkaline earth metals on cobalt-cerium composite oxide catalysts for N2O decomposition[J].Acta Physico-Chimica Sinica,2009,25(6):1033-1039.
[17]Dou Zhe,Feng Ming,Xu Xiufeng.Catalytic decomposition of N2O over Au/Co3O4and Au/ZnC2O4catalysts[J].Journal of Fuel Chemistry & Technology,2013,41(10):1234-1240.
[18]房德仁,刘中民,黎晓琼,等.加料方式对CuO/ZnO/Al2O3系催化剂前驱体性质的影响[J].燃料化学学报,2005,32(6):734-739.
Fang Deren,Liu Zhongmin,Li Xiaoqiong,et al.Influence of precipitation method on the properties of precursors of CuO/ZnO/Al2O3catalysts[J].Journal of Fuel Chemistry & Technology,2005,32(6):734-739.
[19]陈维苗,丁云杰, 薛飞,等.加料方式对共沉淀法制备的Cu/MnO/Al2O3催化剂性能的影响[J].工业催化,2014,22(11):841-846.
Chen Weimiao,Ding YunJie,Xue Fei,et al.Effect of addition modes on catalytic performance of Cu/MnO/Al2O3catalysts prepared by coprecipitation method[J].Industrial Catalysis,2014,22(11):841-846.
[20]高志贤,武建青.铝酸钠溶液碳化制备活化氧化铝[J].燃料化学学报,1998,26(5):468-472.
Gao Zhixian,Wu Jianqing.Preparation of active alumina by carbonation of sodium aluminate solution [J].Journal of Fuel Chemistry & Technology,1998,26(5):468-472.
[21]Wang Y Z,Zhao Y X,Gao C G,et al.Preparation and catalytic performance of Co3O4catalysts for low-temperature CO oxidation[J].Catalysis Letters,2007,116(3/4):136-142.
[22]Voβ M,Borgmann D,Wedler G.Characterization of alumina,silica,and titania supported cobalt catalysts[J].Journal of Catalysis,2002,212(1):10-21.
[23]Xue L,Zhang C,He H,et al.Promotion effect of residual K on the decomposition of N2O over cobalt-cerium mixed oxide catalyst[J].Catalysis Today,2007,126(3):449-455.
Effects of precipitation ways on direct decomposition of N2O over Co3O4catalyst prepared by precipitation method
HuXiaobo,ShiJing,ZhangQiaochu,GuiJiahui,CuiYumeng,WangYongzhao*,ZhaoYongxiang*
(School of Chemistry and Chemical Engineering, Engineering Research Center for Fine Chemicals of Ministry of Education, Shanxi University, Taiyuan 030006, Shanxi, China)
Abstract:Using Co(NO3)2·6H2O as the raw material and K2CO3 as the precipitant,Co3O4 catalysts were prepared by the precipitation method. The structure and properties of the catalysts were characterized by N2-physisorption,XRD,FT-IR,TEM,TPR and ICP.The effect of precipitation ways on the catalytic performance of the catalysts for the direct decomposition of N2O was investigated.The results showed that the precipitation ways had no influence on the textural properties,crystalline phases and particle sizes of Co3O4 catalyst,but significantly affected the amount of residual K and reducibility of Co3O4, which determined the catalytic performance of Co3O4 for direct decomposition of N2O.The amount of residual K on Co-RP catalyst prepared by reverse mode was 1.43wt%,which was higher than that on Co-NP catalyst repared by forward mode.Simultaneously,it was much easier to regenerate the active site (Co(2+)) by donating electrons to Co(3+) over Co-RP catalyst.Under the reaction condition of N2O volume fraction 0.1% and space velocity 10 000 h(-1), Co-RP catalyst for direct decomposition of N2O exhibited better catalytic activity,and the conversion of 100% was obtained at 280 ℃,which was lower 20 ℃ than that over Co-NP catalyst.
Key words:catalyst engineering; Co3O4; precipitation ways; reverse mode; forward mode; N2O decomposition
中图分类号:O633.36;TQ426.6
文献标识码:A
文章编号:1008-1143(2016)01-0041-06
doi:10.3969/j.issn.1008-1143.2016.01.007 10.3969/j.issn.1008-1143.2016.01.007
作者简介:胡晓波,1990年生,山西省忻州市人,在读硕士研究生。
基金项目:国际合作项目(2013DFA40460);山西省科技创新重点团队项目(2012021007)
收稿日期:2015-11-11
催化剂制备与研究
通讯联系人:王永钊,男,副教授,硕士研究生导师;赵永祥,男,教授,博士研究生导师。
