介孔TiO2载体对固体聚合物电解质水电解阳极催化剂性能的影响
2016-04-08米灿根郝传璞宋宇琨
陈 刚, 米灿根, 吕 洪, 郝传璞, 黄 宇, 宋宇琨
(1. 湖南大学材料科学与工程学院, 长沙 410082; 2. 同济大学新能源汽车工程中心, 上海 201804)
介孔TiO2载体对固体聚合物电解质水电解阳极催化剂性能的影响
陈刚1, 米灿根1, 吕洪2, 郝传璞2, 黄宇1, 宋宇琨2
(1. 湖南大学材料科学与工程学院, 长沙 410082; 2. 同济大学新能源汽车工程中心, 上海 201804)
摘要以钛酸四丁酯为原料, 采用溶剂蒸发自组装法(EISA)在不同焙烧温度下制备不同比表面积及结构的介孔TiO2载体. 利用亚当斯熔融法在介孔TiO2载体表面负载IrO2纳米颗粒, 对IrO2/TiO2的结构和性能进行了表征, 并在质子交换膜(PEM)单电池中对IrO2/TiO2催化剂进行了电化学表征. 结果表明, 随着焙烧温度的升高, TiO2载体比表面积降低, 孔径增大, 孔容减小, 组织结构有利于向金红石相转变. TiO2载体的存在明显改善了IrO2颗粒的分布, IrO2晶粒尺寸减小. 在IrO2负载量(质量分数)为40%的情况下, IrO2颗粒易在低比表面积的载体表面形成连续的IrO2导电催化层, 载体比表面积越低其催化活性越高. 在1 A/cm2的电流密度下, IrO2, 40%IrO2/TiO2-2和40%IrO2/TiO2-3催化剂的极化电势分别为2.028, 2.426和2.064 V. 介孔TiO2载体的表面结构及导电性极大影响了催化剂的电化学活性.
关键词氢能; 电解水; 电催化剂载体; 氧化铱; 质子交换膜电解槽
吕洪, 男, 博士, 副教授, 主要从事能源材料等研究. E-mail: lvhong@tongji.edu.cn
随着人们对能源需求的日益增大和石油等传统化石能源的日益枯竭, 寻找新型清洁能源替代化石能源已迫在眉睫. 氢气作为一种能源载体, 由于具有高效、清洁、无污染、来源广泛及便于运输和储存等特点, 被视为未来最理想的能源. 氢能能够有效解决太阳能和风能等间歇性可再生资源发电不稳定等问题[1~3].
固体聚合物电解质(SPE)水电解制氢是以质子交换膜作为电解质的一种制氢技术, 与传统的碱性水电解制氢技术相比, SPE 水电解制氢具有效率高、无污染、安全可靠、可提供高压氢气、结构紧凑以及能量波动适应性强等优点[4]. SPE水电解制氢技术实现产业化所面临的最大挑战来源于其高成本以及高能耗: 现阶段SPE电解槽每电解得到1 m3氢气, 需要耗电(3.9~5.3) kW5h[5]. 在水电解过程中电极的极化损失主要来源于阳极部分, 催化剂和阳极材料是影响SPE水电解性能的关键因素. 在强酸性环境(pH=1~2)下析氧的标准电势为1.23 V(vs. NHE), 电解槽在实际工作中, 阳极槽电压接近2 V(vs. NHE). 在如此苛刻的条件下, 大多数金属电极在达到这一电位前就已溶解, 只有少数贵金属和其氧化物可以稳定存在[6,7]. 因此, 提高阳极材料的催化活性及稳定性, 降低水电解的槽电压, 从而降低成本和提高电解效率是亟待解决的问题.
在强酸和高电位的环境下, IrO2由于具有良好的析氧催化活性、导电性和化学稳定性而成为最好的阳极候选材料[8~10]. 但是IrO2储量少, 且价格昂贵. 利用载体技术[11]开发出合适的催化剂载体, 能够增加多相反应界面和表面活性位, 有效提高催化剂的利用率, 降低贵金属的负载量[12,13], 从而降低SPE电解槽的成本. TiO2在强酸、高电位的环境下具有良好的稳定性, 并已被应用于质子交换膜燃料电池(PEMFC)的阴极催化剂载体的研究中[14]. TiO2作为一种新型载体材料, 其负载的催化剂具有活性高、表面酸性可调、抗中毒性强等优点, 且TiO2本身具有良好的光催化活性[11,15~18]. 本文研究了不同结构TiO2载体对SPE电解槽阳极催化剂IrO2催化活性的影响规律, 并得到了最佳的催化剂载体.
1实验部分
1.1试剂与仪器
嵌段共聚物P123(Mw=5800), Alfa Aesar公司; 钛酸四丁酯(TBT, 质量分数≥98%)、浓盐酸(质量分数40%)、无水乙醇(分析纯)、硝酸钠(分析纯)、氯铱酸(H2IrCl6·6H2O, 分析纯)、异丙醇(分析纯)、H2O2(质量分数30%)和H2SO4(质量分数95%~98%), 国药集团化学试剂有限公司; Nafion溶液(质量分数5%)和Nafion 117质子交换膜, 美国杜邦公司. 40%Pt/C催化剂(JM HiSPEC 4000), 上海河森电气有限公司.
德国Netzsch公司STA449C热分析仪(空气气氛, 测试温度24~1000 ℃, 升温速率5 ℃/min); 德国Bruker公司D8 Discover GADDS X射线衍射仪(XRD, CuKα辐射源, 管电压60 kV, 功率2.2 kW, 步宽0.02°, 扫描速度0.5°/min, 扫描范围15°~90°); 日本Joel 公司JEM-2100F透射电子显微镜; 美国Micrometrics公司ASAP2020比表面积分析仪; 上海辰华仪器有限公司CHI104P电化学工作站; 美国PINE公司AFMS-LXF型旋转圆盘电极. 英国Solartron Mertology公司SI1260型阻抗分析仪.

Fig.1 Structure of the SPE electrolyzer
单电池性能在自制的测试平台上进行, 装置如图1所示. 测试条件为80 ℃恒温去离子水, 流量为40 mL/min.
电化学性能测试条件: 工作电极为涂有催化剂层的玻碳电极, 对电极为铂电极, 参比电极为Ag/AgCl电极, 电解质溶液为0.5 mol/L的H2SO4溶液, 扫描速率为50 mV/s, 扫描范围为-0.17~1.18 V.
1.2介孔二氧化钛载体的制备
采用溶剂蒸发自组装法(EISA)[19,20], 以嵌段共聚物P123为模板剂, 以TBT为前驱体, 制备介孔二氧化钛颗粒.
将TBT与无水乙醇均匀混合, 并于50 ℃剧烈磁力搅拌下滴加浓盐酸与无水乙醇的混合溶液, 滴完后持续剧烈搅拌30 min, 再滴加P123模板剂的无水乙醇溶液, 并持续剧烈搅拌至少2 h, 然后静置形成凝胶. 凝胶组成为n(TBT)∶n(HCl)∶n(H2O)∶n(EtOH)∶n(P123)=1∶1.32∶17∶24∶0.02. 将形成的干凝胶置于马弗炉中, 以1 ℃/min的速度升至100 ℃并保温8 h, 再以1 ℃/min的速度升至200 ℃并保温4 h. 将碳化的样品置于马弗炉中, 在不同温度下焙烧4 h, 焙烧温度为300, 350及450 ℃时所得TiO2载体分别记作TiO2-1, TiO2-2, TiO2-3.
1.3SPE阳极催化剂的制备
采用亚当斯熔融法[21~23]在所制备的TiO2载体表面负载IrO2催化剂. 将催化剂的前驱体氯铱酸溶于异丙醇, 再加入TiO2载体粉末以及研细的硝酸钠粉末球磨4 h, 然后放入预热至350 ℃的马弗炉中保温40 min, 即获得TiO2负载IrO2的催化剂. 活性组分IrO2与载体TiO2的质量比为2∶3. 将利用不同焙烧温度下所得TiO2作为载体制备的负载40%(质量分数)IrO2的催化剂分别标记为40%IrO2/TiO2-1, 40%IrO2/TiO2-2, 40%IrO2/TiO2-3.
1.4膜电极组件(MEA)的制备
以Nafion 117质子交换膜作为固体聚合物电解质. 阴极催化剂为40%(质量分数) Pt/C, 阳极催化剂为上述制备的催化剂样品. 将Nafion 117膜分别用3%(质量分数) H2O2和5%(质量分数) H2SO4溶液浸泡、清洗后, 再用去离子水将残余H2SO4洗净. 采用喷涂法将催化剂浆料分别均匀喷在Nafion 117膜两侧制成膜电极; 其中, 阴极Pt的载量为0.5 mg/cm2, 阳极Ir的载量为2.5 mg/cm2(催化层有效面积为3.645 cm2). 分别采用碳纸和钛网作为阴极和阳极的集电极, 流场板采用金属钛板, 再将其组装于单电池电解槽进行测试.
2结果与讨论
2.1TG-DSC分析
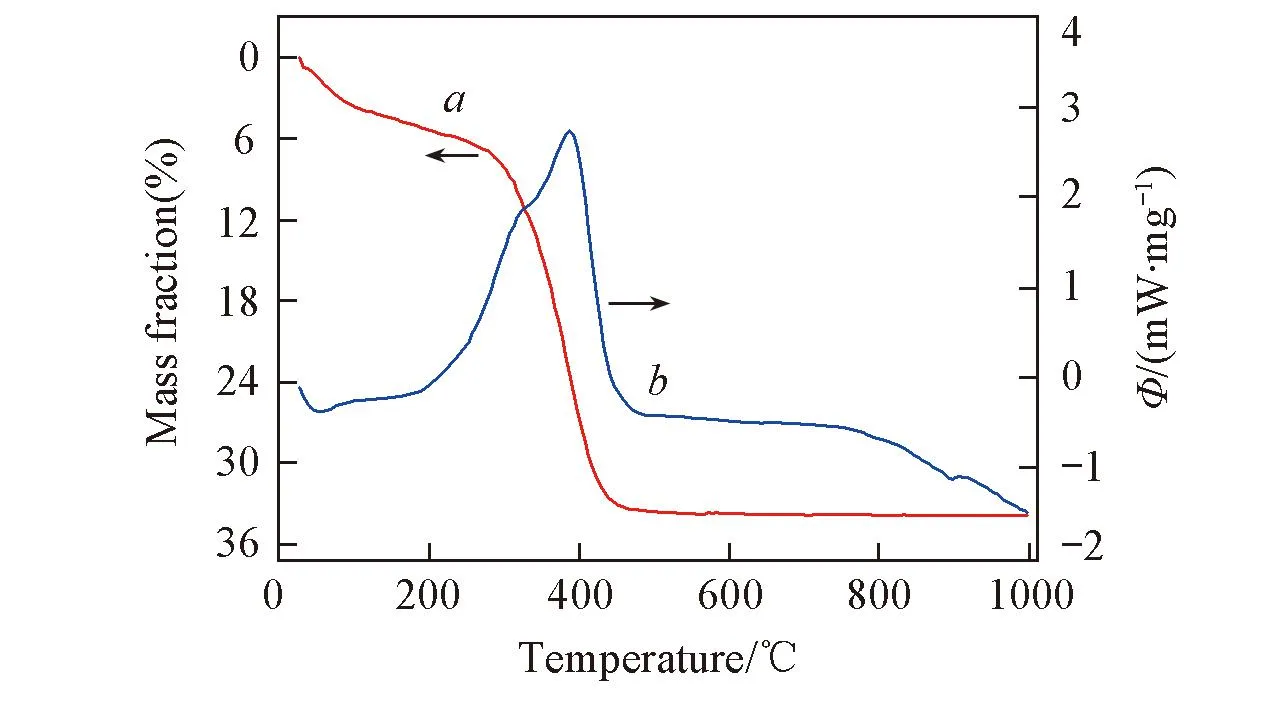
Fig.2 TGA(a) and DSC(b) curves of TiO2 before calcination
图2为含模板剂的碳化后载体样品的TG-DSC分析结果. 可以看出, 在温度低于200 ℃之前质量损失很少, 此部分失重主要是水分的蒸发所致; 当温度达到250 ℃以后, 样品质量快速下降, 直至温度达到450 ℃, 样品质量保持不变, 此过程的失重主要是模板剂的燃烧分解导致的. 在380 ℃左右出现一个明显的放热峰, 表明体系中的模板剂开始燃烧分解, 系统放热; 直至温度达到450 ℃后无明显变化; 当温度达到750 ℃时系统开始有明显的吸热现象, 表明从该温度开始, TiO2发生由锐钛矿相向金红石相的转变.
2.2微观结构特性分析
表1为不同焙烧温度下TiO2的比表面积(S{BET)、孔径和孔容(Vp)结果. 可以看出, 随着焙烧温度的升高, 样品的比表面积迅速下降, 孔径增大, 孔容逐渐减小. 这主要是由于焙烧温度的升高使得孔道结构坍塌破坏造成的. 与300 ℃下焙烧所得样品相比, 350 ℃下焙烧所得样品的比表面积和孔容减小幅度较小, 这是因为焙烧温度相差较小, 仅仅使小部分孔道结构被破坏. 而在450 ℃下焙烧的样品比表面积下降幅度大, 孔径增大约1.6倍, 孔容几乎为0, 这是由于焙烧温度过高导致模板剂去除得太快, 失去模板剂支撑的多孔结构大部分迅速坍塌, 从而造成比表面积迅速下降, 孔径增大, 孔容减小. 上述结果表明, 载体的表面结构对焙烧温度非常敏感, 焙烧温度决定着载体的表面结构.

Table 1 BET parameters of calcined TiO2 samples
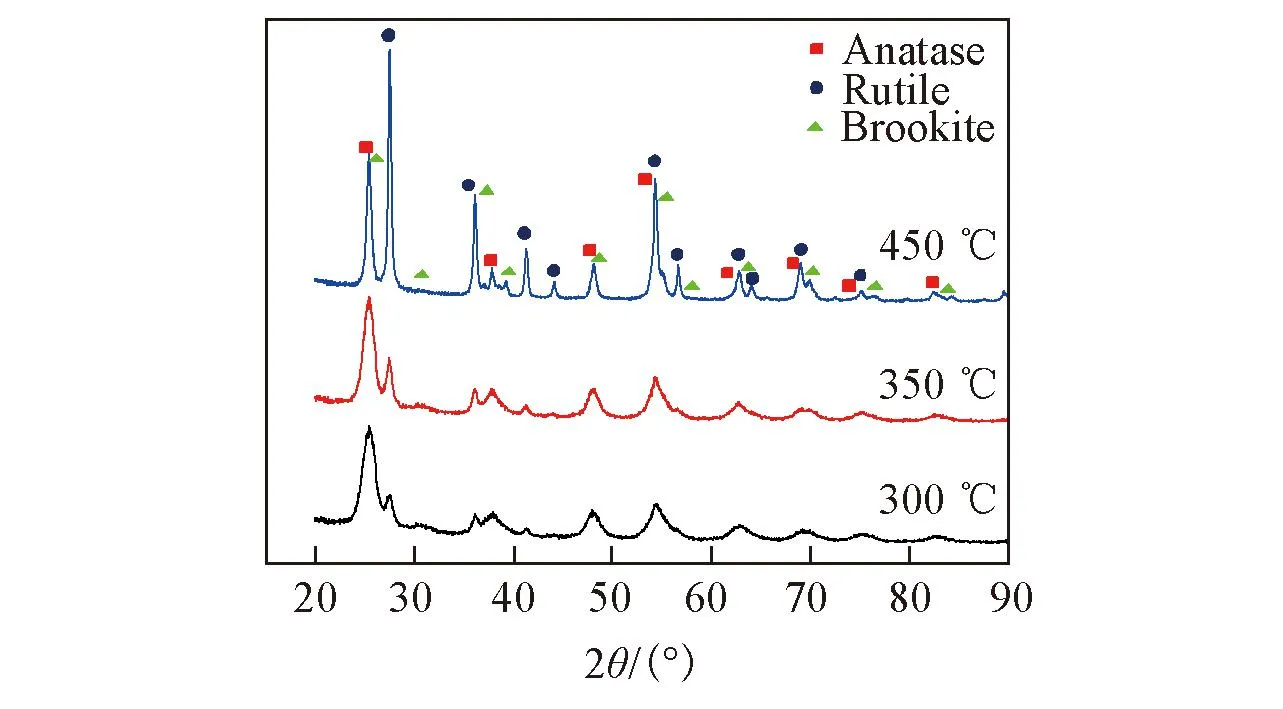
Fig.3 XRD patterns of TiO2 samples calcined at different temperatures

Fig.4 XRD patterns of pure IrO2(a) and 40%IrO2/TiO2-1(b), 40%IrO2/TiO2-2(c) and 40%IrO2/TiO2-3(d) catalysts
不同焙烧温度下所得TiO2载体的XRD谱如图3所示. 可以看出, 所制备的TiO2为金红石相、锐钛矿相以及板钛矿相的混合相, 其中主要为金红石相和锐钛矿相. 随着TiO2样品焙烧温度的升高, 金红石相的衍射峰显著增强, 表明金红石相含量增大. 这说明焙烧温度越高越有利于锐钛矿相向金红石相的转变. 焙烧温度的升高使得各特征峰的强度不断增强, 300和350 ℃焙烧温度下所得TiO2的衍射峰强度和宽度相差不大, 450 ℃焙烧温度下所得TiO2的衍射峰强度明显增大, 衍射峰宽度显著变窄, 根据谢乐公式[24]计算得出: TiO2-1, TiO2-2和TiO2-3平均晶粒尺寸分别为6.5, 8.4, 16.5 nm. 表明随着焙烧温度的升高, TiO2结晶程度不断提高, 同时晶粒不断长大.
图4为不同焙烧温度下制备的TiO2负载IrO2后的XRD谱图. 可见, IrO2的负载使得TiO2的各个衍射峰强度明显降低, 这是由于催化剂粒子在载体表面均匀分散所致. 此外, 谱图中IrO2的衍射峰很少且不明显, 这是由于反应温度较低导致IrO2颗粒结晶不完善所致. 通过对比发现, 40%IrO2/TiO2-2中金红石相的特征峰强度较弱, 其金红石相的含量较低, 金红石晶粒尺寸相对较小; 40%IrO2/TiO2-3中金红石相含量较高.
对比图3和图4可见, 反应过程中未产生IrO2和TiO2的化合物, 表明IrO2和TiO2具有良好的热化学相容性; 同时发现负载IrO2的TiO2的衍射峰产生一定的宽化, 这是由于IrO2晶粒细小, 衍射峰较宽, 部分衍射峰与TiO2的衍射峰距离较近, 产生叠加效果, 导致TiO2的衍射峰变宽.
图5为不同焙烧温度下所得TiO2载体的TEM照片, 可见, TiO2-1和TiO2-2的晶粒尺寸较均匀, 晶粒堆垛结合形成了介孔结构, 此形貌与Soler-Illia[20]解释EISA法成型机理的“纳米积木”模型相吻合; 而TiO2-3的介孔结构则不明显, 这是由焙烧温度过高导致介孔坍塌所致, 与BET分析结果一致. TEM结果显示, TiO2-1晶粒尺寸集中分布在4~6 nm, 且晶格条纹不明显; TiO2-2晶粒尺寸集中分布在7~9 nm, 且晶格条纹清晰; TiO2-3晶粒尺寸集中分布在15~18 nm, 且晶格条纹最清晰. 表明随着焙烧温度升高, TiO2结晶度提高, 晶粒平均粒径增大, 这与XRD分析结果相吻合. 通过对不同焙烧温度下所得TiO2的微观结构分析可见, 350 ℃焙烧温度下可以获得结晶度良好、晶粒尺寸和比表面积合适的介孔TiO2.

Fig.5 TEM images of TiO2 supports calcined at different temperatures (A1), (A2) TiO2-1; (B1), (B2) TiO2-2; (C1), (C2) TiO2-3.

Fig.6 TEM images of pure IrO2 and 40% IrO2/TiO2 catalysts(A1, A2) IrO2; (B1, B2) 40%IrO2/TiO2-1; (C1, C2) 40%IrO2/TiO2-2; (D1, D2) 40%IrO2/TiO2-3.
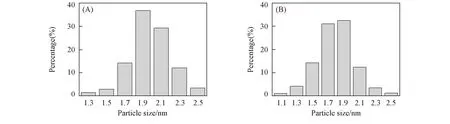
Fig.7 Particles size distribution of pure IrO2 catalyst(A) and IrO2 particles size distribution of 40%IrO2/TiO2-2(B)
图6为以亚当斯熔融法制备的纯IrO2, 40%IrO2/TiO2-1, 40%IrO2/TiO2-2及40%IrO2/TiO2-3样品的TEM照片. 由图6可见, 无载体IrO2样品的颗粒团聚严重, 催化剂晶粒尺寸较大; 40%IrO2/TiO2-1上的IrO2不连续分布在TiO2晶粒表面; 40%IrO2/TiO2-2上的催化剂晶粒在载体表面分布最均匀, 但彼此之间并未形成连续网络; 40%IrO2/TiO2-3中连续均匀的IrO2催化层包裹在TiO2晶粒的外表面, 形成类似以TiO2为核, 连续IrO2层为壳的结构, 但是仍有一小部分催化剂晶粒单独团聚在一起而没有负载于载体上. 表明具有介孔结构的TiO2载体可以有效改变IrO2晶粒的分散形态, 提高其分散性. 介孔分布越均匀, 比表面积越大, 可为IrO2生长过程提供的有效形核点就越多, 从而细化IrO2晶粒, 有效改善IrO2颗粒的分布, 减少团聚现象.
通过对各个样品中IrO2晶粒尺寸进行统计计算, 得到晶粒粒径分布图, 如图7所示. 可见, 纯IrO2晶粒粒径主要集中在1.7~2.3 nm, 其平均晶粒尺寸约为2.0 nm; 40%IrO2/TiO2上的IrO2晶粒粒径主要集中在1.5~2.1 nm, 其平均晶粒尺寸约为1.8 nm. 这说明载体的存在有利于形成更细小的IrO2颗粒, 其主要原因是载体的存在使得催化剂粒子在反应过程中依附于载体的形核点发生异质形核, 增加形核率, 降低形核所需能量, 从而使得IrO2晶粒尺寸更小.
2.3催化剂的电化学性能
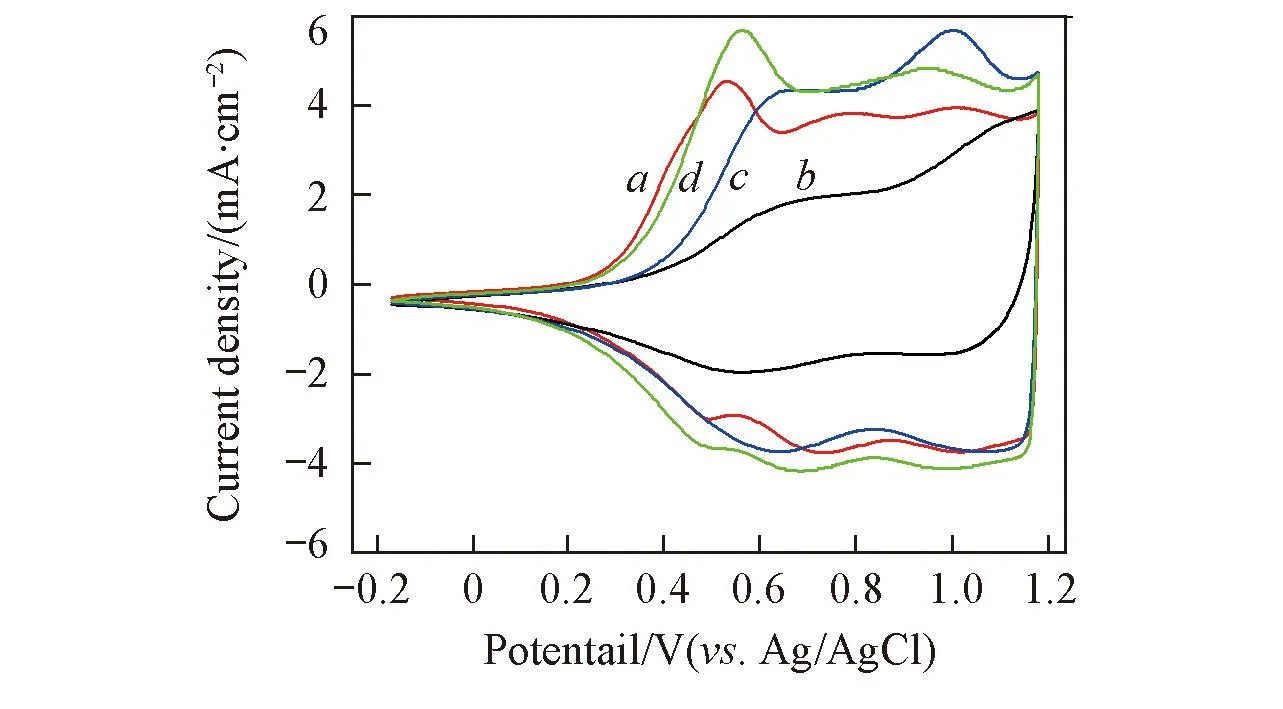
Fig.8 CV curves of catalyst samples in N2-satura-ted 0.5 mol/L H2SO4 solution at room temperature(scan rate: 50 mV/s)a. IrO2; b. 40%IrO2/TiO2-1; c. 40%IrO2/TiO2-2; d. 40%IrO2/TiO2-3.
图8为各催化剂样品在0.5 mol/L H2SO4溶液中的循环伏安曲线. 在施加电位扫描过程中, IrO2中Ir的价态会发生改变并伴随着明显的电流峰值变化. 图8中IrO2与40%IrO2/TiO2-3的CV曲线形状几乎一致, 这说明它们发生了相同的氧化还原反应. 2个析氧峰分别发生在0.50和0.97 V处, 其中0.5 V处的峰更加显著. 40%IrO2/TiO2-1的伏安曲线上2个析氧峰分别发生在0.60和0.97 V处, 其中0.97 V处的峰更加显著. 根据IrO2在酸性介质中的析氧反应机理, 在约0.5 V电位下, 主要是发生IrⅢ/IrⅣ氧化还原反应, IrⅣ/IrⅥ的氧化还原反应发生在约0.97 V电位下, 其具体转化过程[25~28]为

(1)
当x=y=z=1时, 发生IrⅢ/IrⅣ价态转变;x=1,y=z=2时, 发生IrⅣ/IrⅥ价态转变[28].
实验结果表明, 与40%IrO2/TiO2-2相比, IrO2与40%IrO2/TiO2-3中铱的价态转变主要发生在较低电位下, 表明氧的吸附主要在低电位进行, 因此, 更利于析氧反应的发生, 从而获得更好的催化活性. 结合XRD图谱分析, TiO2-3中金红石相的峰强度明显高于TiO2-2和TiO2-1, 其金红石相含量最高, 因此, 金红石相TiO2有利于促进IrO2在低电位下进行氧的吸脱附反应. 同时, 具有多孔结构的TiO2载体可以扩展三相界面, 提高传质的效率, 同时有利于反应过程中产生的氧气排出, 从而产生协同效应, 提高催化剂的活性.
通常用伏安电荷量q*表征催化剂颗粒与溶液进行质子交换的有效活性位点数, 并以此判断催化剂的电化学活性面积[28,29]. 对图8中的CV曲线进行积分计算得出IrO2, 40%IrO2/TiO2-1, 40%IrO2/TiO2-2和40%IrO2/TiO2-3样品的伏安电荷量大小分别为1264, 665, 1304及1472 mC/(cm2·mg), 催化活性顺序为40%IrO2/TiO2-3>40%IrO2/TiO2-2>IrO2>40%IrO2/TiO2-1. 根据Mazúr等[15]的研究成果, 载体比表面积在100 m2/g以上时, 40%的催化剂担载量不足以在载体表面形成连续的IrO2聚集薄层, 因此, 对于样品40%IrO2/TiO2-1和40%IrO2/TiO2-2, 由于TiO2导电性差, 且TiO2载体孔容大, 比表面积高, 导致催化剂粒子过于分散, IrO2粒子塞积在介孔结构中被阻隔, 相互之间不能够良好接触, 形成有效的导电网络, 反应活性位减少, 从而导致催化剂的催化活性不高. 对于40%IrO2/TiO2-3, 虽然其比表面积较低, 孔径大, 但是有利于催化剂晶粒在载体表面覆盖, 将载体完全包裹, 形成连续的IrO2薄膜层. 所以40%IrO2/TiO2-3的电催化活性最好. 无载体IrO2由于颗粒团聚严重, 只有部分催化剂起到了催化作用, 裹在内部的催化剂粒子只起到导电效果并没有参与析氧反应过程, 因而IrO2有效利用率低, 催化活性受到限制.
2.4阳极催化剂单电池测试分析
图9为使用不同阳极催化剂的单电解池极化曲线. 在1.0 A/cm2的电流密度下, 40%IrO2/TiO2-2过电位为2.426 V; 40%IrO2/TiO2-3的过电位为2.064 V; 纯IrO2的过电位为2.024 V. Mazúr等[15]以商业的TiO2(SBET=50 m2/g) 为载体, 负载60%IrO2催化剂, 在90 ℃和0.8 A/cm2电流密度下测得复合催化剂的过电位为1.8 V; 本文中40%IrO2/TiO2-3在80 ℃, 0.8 A/cm2电流密度下的过电位为1.9 V. 由于IrO2担载量和温度对过电位的影响非常显著, 高的IrO2的担载量和温度都会降低电解的过电位, 提高单电解池性能, 因此, 判断本文制备的复合催化剂具有更好的催化活性.

Fig.9 Polarization curves of catalysts at 80 ℃ for the single cell PEM electrolyzera. Pure-IrO2; b. 40%IrO2/TiO2-2; c. 40%IrO2/TiO2-3.
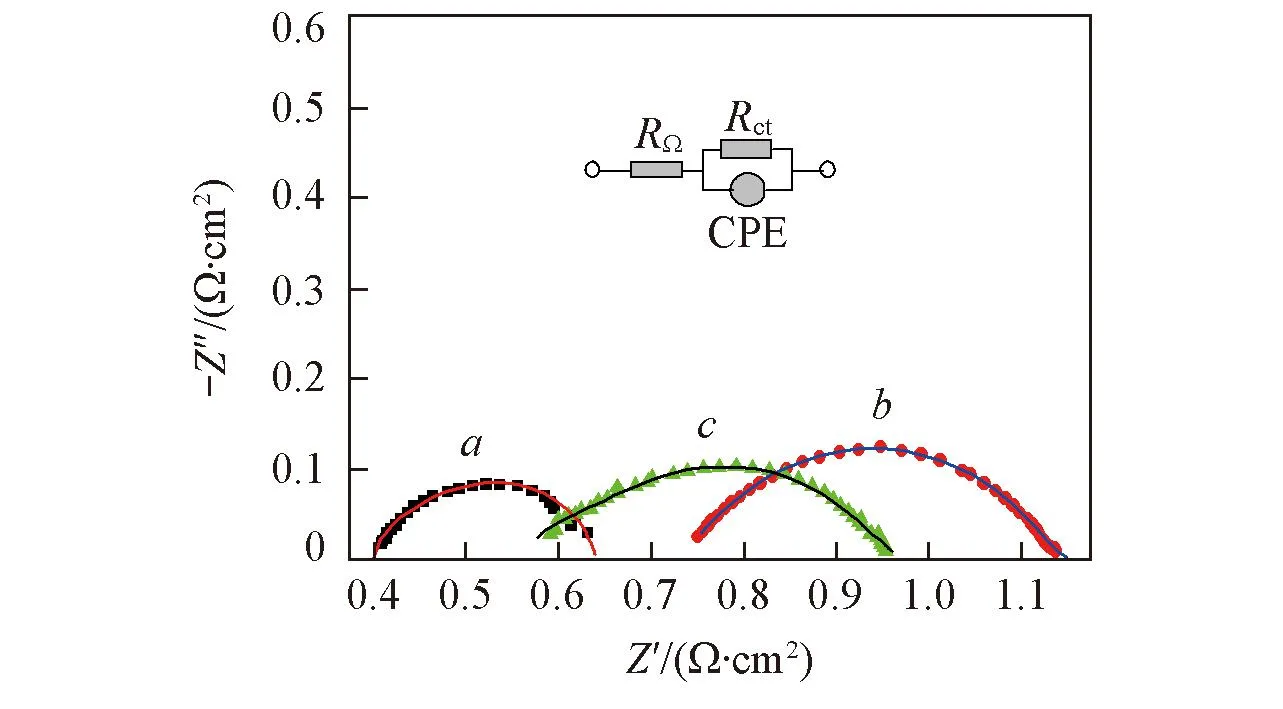
Fig.10 Nyquist diagram of catalyst samples in the PEM single cell a. IrO2 fit result; b. 40%IrO2/TiO2-2 fit result; c. 40%IrO2/TiO2-3 fit result.
图10为在0.1 A/cm2下测得的Nyquist图, 其中实线为用Zview软件拟合所得曲线, 插图为模拟等效电路图, 图中RΩ表示SPE电解池的整体欧姆阻抗, 主要包含Nafion膜、催化剂层、两极板、钛网以及导线的欧姆电阻,Rct表示氧化物与溶液界面之间产生的电荷传递阻抗, CPE为常相位角元件. 表2列出了拟合后所得阻抗参数.
由表2可见, 纯IrO2的欧姆阻抗为0.40 Ω·cm2, 40%IrO2/TiO2-2和40%IrO2/TiO2-3欧姆阻抗分别是0.74和0.57 Ω·cm2. 这是由于IrO2具有优良的导电性, TiO2导电性差, 因此, 纯IrO2的欧姆阻

Table 2 Impendence parameters obtained from fitting the experimental data to the RΩ(RctCPE)
*RΩ: Ohmic resistance; CPE: constant phase element;Rct: charge-transfer resistance; n: index of CPE.
抗低于IrO2/TiO2复合催化剂. 另外, 40%IrO2/TiO2-3在载体表面形成了连续的IrO2导电催化层, 因而其欧姆阻抗比40%IrO2/TiO2-2低. 40%IrO2/TiO2-2与40%IrO2/TiO2-3催化剂的电荷传递阻抗相当, 分别为0.400和0.385 Ω·cm2. 而无载体的IrO2催化剂的电荷传递阻抗为0.240 Ω·cm2. 这主要是由于IrO2颗粒在载体表面均匀分散, 造成彼此之间没有形成完全有效的导电网络, 电荷在氧化物之间传递的距离增大, 从而增大了Rct. 此外, 在0.1 A/cm2电流密度下, 纯IrO2的CPE值为143 mF/cm2, 40%IrO2/TiO2-2和40%IrO2/TiO2-3的CPE值分别为102和117 mF/cm2. CPE值反映了有效活性表面积的大小. 在低电流密度条件下, 有效活性面积主要与IrO2的担载量相关, 因此, 纯IrO2的CPE值最大; 40%IrO2/TiO2-2由于载体比表面积较大, 孔容较大, 包裹到介孔中的部分IrO2颗粒的活性面积被遮挡, 活性面积减少, 使得其CPE值减小.
3结论
采用溶剂挥发诱导自组装法制备了介孔TiO2载体, 其比表面积可高达172 m2/g. 焙烧温度对介孔TiO2的表面结构影响显著: 焙烧温度越高, 其比表面积越低, 孔道破坏越严重, 孔径越大; 同时焙烧温度的升高有助于金红石相的生成并促进晶粒的生长. 采用亚当斯融化法可制备晶粒尺寸小于2 nm的IrO2晶粒, TiO2载体的存在有利于改善IrO2催化剂颗粒的分布, 并细化晶粒尺寸, 从而提高IrO2的催化活性面积. 在1.0 A/cm2的电流密度下, 40%IrO2/TiO2-3催化剂的极化电势可达2.064 V, 与纯IrO2性能相当. 载体中金红石相的存在有利于产生协同效应, 提升催化剂的析氧反应活性.
参考文献
[1]Troncoso E., Newborough M.,Int.J.HydrogenEnergy, 2011, 36(1), 120—134
[2]Ipsakis D., Voutetakis S., Seferlis P., Stergiopoulos F., Elmasides C.,Int.J.HydrogenEnergy, 2009, 34(16), 7081—7095
[3]Barbir F.,Sol.Energy, 2005, 78(5), 661—669
[4]Lu P. W. T., Srinivasan S.,J.Appl.Electro.Chem., 1979, 9(3), 269—283
[5]Holladay J. D., Hu J., King D. L., Wang Y.,Catal.Today, 2009, 139(4), 244—260
[6]Andolfatto F., Durand R., Michas A., Millet P., Stevens P.,Int.J.HydrogenEnergy, 1994, 19(5), 421—427
[7]Grigoriev S. A., Porembsky V. I., Fateev V. N.,Int.J.HydrogenEnergy, 2006, 31(2), 171—175
[8]Siracusano S., Baglio V., Di Blasi A., Briguglio N., Stassi A., Ornelas R., Arico A. S.,Int.J.HydrogenEnergy, 2010, 35(11), 5558—5568
[9]Da Silva L. M., Boodts J. F. C., DeFaria L. A.,ElectrochimicaActa, 2000, 45(17), 2719—2727
[10]Kötz R., Stucki S.,ElectrochimcaActa, 1986, 31(10), 1311—1316
[11]Mamaca N., Mayousse E., Arrii-Clacens S., Napporn T. W., Servat K., Guillet N., Kokoh K. B.,Appl.Catal.B-Environ., 2012, 111, 376—380
[12]Song S., Zhang H., Ma X., Shao Z., Baker R. T., Yi B.,Int.J.HydrogenEnergy, 2008, 33(19), 4955—4961
[13]Siracusano S., Baglio V., Stassi A., Ornelas R., Antonucci V., Aricò A. S.,Int.J.HydrogenEnergy, 2011, 36(13), 7822—7831
[14]Huang S. Y., Ganesan P., Park S., Popov B. N.,J.Am.Chem.Soc., 2009, 131(39), 13898—13899
[15]Mazúr P., Polonsky J., Paidar M., Bouzek K.,Int.J.HydrogenEnergy, 2012, 37(17), 12081—12088
[16]Zhang R., Elzatahry A. A., Al-Deyab S. S., Zhao D.,NanoToday, 2012, 7(4), 344—366
[17]Gao W., Wu F. Q., Luo Z., Fu J. X., Wang D. J., Xu B. K.,Chem.J.ChineseUniversities, 2001, 22(4), 660—662(高伟, 吴凤清, 罗臻, 富菊霞, 王德军, 徐宝琨. 高等学校化学学报, 2001, 22(4), 660—662)
[18]Wang J. Q., Xin B. F., Yu H. T., Ren Z. Y., Qu P. F., Fu H. G.,Chem.J.ChineseUniversities, 2003, 24(6), 1093—1096(王建强, 辛柏福, 于海涛, 任志宇, 曲鹏飞, 付宏刚. 高等学校化学学报, 2003, 24(6), 1093—1096)
[19]Grosso D., Cagnol F., Soler-Illia G. D. A., Crepaldi E. L., Amenitsch H., Brunet-Bruneau A., Sanchez C.,Adv.Funct.Mater., 2004, 14(4), 309—322
[20]Soler-Illia G. J. A. A., Louis A., Sanchez C.,Chem.Mater., 2002, 14(2), 750—759
[21]Adams R., Voorhees V., Shriner R. L.,Org.Synth., 1941, 92
[22]Rasten E., Hagen G., Tunold R.,ElectrochimicaActa, 2003, 48(25), 3945—3952
[23]Cheng J., Zhang H., Ma H., Zhong H., Zou Y.,Int.J.HydrogenEnergy, 2009, 34(16), 6609—6613
[24]Birks L. S., Friedman H.,J.Appl.Phys., 1946, 17(8), 687—692
[25]Kong F. D., Zhang S., Yin G. P., Wang Z. B., Du C. Y., Chen G. Y., Zhang N.,Int.J.HydrogenEnergy, 2012, 37(1), 59—67
[26]Li G., Yu H., Wang X., Sun S., Li Y., Shao Z., Yi B.,Phys.Chem.Chem.Phys., 2013, 15(8), 2858—2866
[27]Marshall A., Børresen B., Hagen G., Sunde S., Tsypkin M., Tunold R.,Russ.J.Electrochem., 2006, 42(10), 1134—1140
[28]Puthiyapura V. K., Mamlouk M., Pasupathi S., Pollet B. G.,J.PowerSources, 2014, 269, 451—460
[29]Fachinotti E., Guerrini E., Tavares A. C., Trasatti S.,J.Electroanal.Chem., 2007, 600(1), 103—112
Mesoporous TiO2as the Anode Catalyst Support for Solid
Polymer Electrolyte Water Electrolysis†
CHEN Gang1*, MI Cangen1, LÜ Hong2*, HAO Chuanpu2, HUANG Yu1, SONG Yukun2
(1.CollegeofMaterialsScienceandEngineering,HunanUniversity,Changsha410082,China;
2.CleanEnergyAutomotiveEngineeringCenter,TongjiUniversity,Shanghai201804,China)
AbstractTiO2support was prepared through evaporation-induced self-assembly(EISA) by using butyl titanate as starting material. Three TiO2samples were calcined at different temperatures and then IrO2was loaded on them with a mass ratio of IrO2/TiO2of 2/3 by a modified Adams fusion method. The samples were characterized by X-ray diffraction(XRD), specific surface area measurement, thermogravimetric and differential scanning calorimetry analysis(TGA-DSC), transmission electron microscopy(TEM) and electrochemical testing. The electrochemical activity of the catalysts was investigated in a single cell proton exchange membrane(PEM) electrolyzer consisting of a Pt/C cathode and a Nafion117 membrane. The results suggested that as the calcination temperature increased, the mesoporous structure of TiO2was destroyed, the pore size increased and the pore volume reduced, and the phase of TiO2transferred from anatase to rutile structure. Utilization of the TiO2support resulted in a reduction in the size of the IrO2crystallites and improved the distribution of catalyst. It was found that the lower the specific surface area of the support was, the higher the electrochemical activity of the catalyst was. This is most likely due to the formation of a conductive IrO2film on the surface of non-conductive supports with 40%(mass fraction) loading of IrO2. The IrO2, 40%IrO2/TiO2-2 and 40%IrO2/TiO2-3 catalysts showed a polarization potential of 2.024, 2.426 and 2.064 V, respectively, under a current density of 1.0 A/cm2. These results suggest that the surface structure of the support has a great influence on the catalytic activity of IrO2.
KeywordsHydrogen; Water electrolysis; Electrocatalyst support; IrO2; Proton exchange membrane electrolyzer
(Ed.: S, Z, M)
† Supported by the National Natural Science Foundation of China(No.21306141).
doi:10.7503/cjcu20150389
基金项目:国家自然科学基金(批准号: 21306141)资助.
收稿日期:2015-06-04. 网络出版日期: 2015-12-20.
中图分类号O643
文献标志码A
联系人简介:陈刚, 男, 博士, 教授, 主要从事金属材料及功能材料研究. E-mail: chengang@hnu.edu.cn
