硼酸化合物在药物发现方面的研究进展
2016-03-21吴忠玉姚庆强孙敬勇
王 凯,吴忠玉,姚庆强*,孙敬勇
( 1.山东省医学科学院药物研究所,山东济南250062; 2.济南大学-山东省医学科学院医学与生命科学学院,山东济南250062; 3.山东省罕少见病重点实验室,山东济南250062)
硼酸化合物在药物发现方面的研究进展
王凯1,2,3,吴忠玉1,2,3,姚庆强1,2,3*,孙敬勇1,2,3
( 1.山东省医学科学院药物研究所,山东济南250062; 2.济南大学-山东省医学科学院医学与生命科学学院,山东济南250062; 3.山东省罕少见病重点实验室,山东济南250062)
摘要:硼酸化合物广泛应用于有机合成、荧光探针以及药物发现等方面.尤其是随着硼替佐米被美国FDA批准上市,硼酸类化合物在药物发现方面的前景引起了科学家的极大兴趣.本文综述了硼酸化合物在药物发现方面,尤其是作为酶抑制剂和特异性配体等方向的最新研究进展.
关键词:硼酸化合物;硼替佐米;酶抑制剂;药物发现
目前,硼酸化合物被广泛应用于有机合成、碳水化合物的识别以及药物发现.在有机化学领域,硼酸化合物广泛应用于二元醇保护、氨基酸不对称合成、Diels-Alder反应、Suzuki偶联反应等方面.在药物发现方面,硼酸化合物可作为酶抑制剂或特异性配体,用于肿瘤和微生物感染等疾病的治疗[1-3].另外,其还可作为用于识别过氧化氢、糖类、铜离子、氟离子和儿茶酚胺类等物质的荧光探针[4].
20世纪70年代科学家第一次开发硼酸,得到了β-内酰胺酶和胰凝乳蛋白酶的酶抑制剂[5-6].经过科学家潜心研究,许多具有强效亲和力的硼酸类抑制剂被相继合成并报道[7-9].尤其是2003年,硼酸类蛋白酶体抑制剂硼替佐米的成功上市[10],极大地提高了科学家在硼酸类药物研发领域的兴趣和信心.本文作者将对硼酸类的蛋白酶体抑制剂、丝氨酸蛋白酶体抑制剂、组蛋白去乙酰化酶抑制剂、RNA聚合酶抑制剂、CXCR1和CXCR2的非竞争性硼酸抑制剂等在近十年的研究进行总结和讨论.
1 有机硼酸类酶抑制剂主要作用机制
硼原子的价电子结构是2s22p1,原子结构中拥有一个空轨道.硼原子以sp2杂化形成共价分子,余下的一个空轨道可以作为路易斯酸,接受外来的孤对电子,形成sp3杂化的四面体构型的配合物.在水溶液里,H2O分子中氧原子的孤对电子进入硼原子的空轨道,释放一个质子产生sp3杂化的硼(正四面体型,图1).由于硼酸化合物pKa较低,在生理pH条件下,硼原子以sp3杂化形式存在.这种四面体分子构型模拟了酶催化状态下四面体的过渡状态,从而竞争性地抑制了酶的活性[11-13].
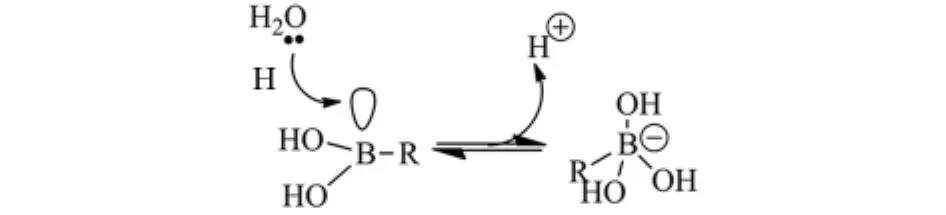
图1 sp2到sp3杂化类型的转化Fig.1 Transformation of sp2to sp3hybridized forms
2 蛋白酶体抑制剂
蛋白酶体是一种巨型蛋白质复合物,广泛存在于真核生物、古菌以及一些原核生物中.在真核生物中,蛋白酶体位于细胞核和细胞质中,主要作用是降解细胞非必需的或受到损伤的蛋白质.作为一个多重催化性的苏氨酸蛋白酶,蛋白体酶在泛激素依赖型细胞质降解中起着重要的作用[14-16].蛋白酶体抑制剂通过抑制蛋白酶体的活性,从而影响细胞生长所需蛋白、细胞因子和信号分子的传导,干扰了细胞正常的增殖、分化和凋亡过程,对肿瘤细胞的抑制更为明显.
近年来,蛋白酶体抑制剂在临床抗癌方面效果显著.2003年,硼替佐米( 1,PS-341,万珂)成为第一个被FDA批准上市的蛋白酶体抑制剂,主要用于治疗多发性骨髓瘤( MM).同时也用于治疗巨球蛋白血症的巨球蛋白血( WM)以及非霍奇金淋巴瘤( NHL)[17].

图2 化合物1,2,3的分子结构Fig.2 Molecular structure of compound 1,2 and 3
科学家正在寻找第二代蛋白酶体抑制剂,以减轻外周神经病变的副作用,提高对多发性骨髓瘤病人的疗效[18-19].目前,两种硼酸类蛋白酶体抑制剂正在进行临床试验.Ixazomib( 2,MLN9708)是用于治疗MM的口服型蛋白酶抑制剂,成为第一个进入Ⅱ期临床试验的药物.化合物3( CEP-118770)在抑制MM的小鼠异种移植模型中表现出良好的抗肿瘤活性,目前已经进入Ⅰ期临床试验.

图3 化合物4的分子结构和IC50值Fig.3 Molecular structure and IC50value of compound 4
WATANABLE等[20]使用化合物4 ( TP-110,图3 )作为先导化合物合成了一系列Tyropeptin硼酸类化合物.构效关系( SAR)研究表明,通过使用硼酸基团代替醛基,表现出更强的糜蛋白酶体抑制活性.在N端去掉酰基后活性降低,说明酰基是保持蛋白酶体抑制性的必要官能团.进一步SAR试验,采用不同的取代基替换位于N端的R基,其中引入1-萘基乙酰( 5a和5b)和3-苯氧基苯乙酰( 5c)具有较高的蛋白酶体抑制活性.另外,在R1位引入异丙基的活性明显高于引入甲氧基苯基所得的衍生物.

图4 化合物5a,5b和5c的分子结构和IC50值Fig.4 Molecular structure and IC50value of compound 5a,5b and 5c
CUI和LI等[21]根据3D-QSAR研究了三肽衍生物,获得了一系列三肽硼酸类蛋白酶体抑制剂.虽然这些新型化合物的生物学结果与3D-QSAR模型的预测值并不一致,但发现了一系列结构新颖的抑制剂.特别是化合物6的活性比硼替佐米高20%.之后,ZHU等[21]报道了72个二肽硼酸类蛋白酶体抑制剂,并对构效关系进行了研究.研究表明化合物7在蛋白酶体抑制方面和血液肿瘤细胞株细胞毒性(分别为IC50小于2 nmol/L和IC50小于1 μmol/ L)方面拥有很强的活性.药代动力学曲线证明化合物7比硼替佐米拥有更长的半衰期和更高的血浆浓度.

图5 化合物6和7的分子结构Fig.5 Molecular structure of compound 6 and 7
3 丝氨酸蛋白酶抑制剂
丝氨酸蛋白酶是一种广泛存在于真核生物和原核生物中的蛋白水解酶.根据丝氨酸蛋白酶的结构不同可以分成两类:枯草杆菌蛋白酶类和胰凝乳蛋白酶类[22].它们负责协调不同的生理功能,包括凝血、消化、复制和免疫反应功能.抑制丝氨酸蛋白酶可以影响一系列重要的生理过程,包括血液凝结、免疫反应、炎症和肿瘤等.硼酸化合物通过模拟缩氨酸水解过程的四面体过渡状态,竞争性地抑制丝氨酸蛋白酶(图6).下面分别介绍凝血酶抑制剂、β-内酰胺酶抑制剂、中丝氨酸蛋白酶抑制剂、DPP-IV抑制剂等硼酸类丝氨酸蛋白酶抑制剂的研究进展.
3.1凝血酶抑制剂
血栓栓塞类疾病是致死和致残的主要因素.凝血因子Ⅻa和凝血酶可以激活凝血因子Ⅺ,凝血因子Ⅺ激活凝血因子Ⅺa.凝血因子Ⅺa可以促进血液凝固,从而增加了静脉血栓的风险.目前,科学家在临床研究中发现了包括候选化合物在内的凝血酶和凝血因子Ⅺa抑制剂[15-16,23].2006年,LAZAROVA等[24]合成了一系列苯基硼酸类化合物,特别是硼酸酯8拥有良好的凝血酶抑制性.该化合物在微摩尔浓度下可有效抑制凝血因子Ⅺa,但是在针对胰蛋白酶( IC50>200 μmol/L)、凝血酶( IC50=12.3 μmol/L)和Ⅹa因素( IC50=43.6 μmol/L)时抑制活性较弱.

图7 化合物8的分子结构Fig.7 Molecular structure of compound 8
3.2 β-内酰胺酶抑制剂
3.2.1烷基硼酸
β-内酰胺酶催化抗生素β-内酰胺键的水解,使β-内酰胺环裂解,从而使β-内酰胺类抗生素失效,比如青霉素和头孢菌素.β-内酰胺酶被分成许多不同的种类,其中A类β-内酰胺酶(比如基于质粒的TEM青霉素酶)以及C类β-内酰胺酶(比如AmpC-β-内酰胺酶)的研究具有重要的临床意义[25-26].硼酸化合物在生理状态下,其四面体构型模拟了β-内酰胺酶水解过程的过渡态结构(图8)[27].
通过增加硼酸化合物与β-内酰胺头孢菌素的相似度,将会显著增强对β-内酰胺酶的抑制性.PRATI等[28]利用化合物9作为先导物,通过结构优化获得了一系列高效的硼酸类β-内酰胺酶抑制剂.其中,含有双键的衍生物10针对AmpC-β-内酰胺酶时抑制活性较强( Ki= l nmol/L).另外,利用生物电子等排体原理,用四氮唑代替羧基得到了具有微弱抑制性的化合物11.同时,内酰胺酶和化合物10的晶体结构证实硼酸化合物为四面体的几何构型,其中关键的结构类似于在β-内酰胺结构观察到的过渡态类似物.
2013年BONOMO等[29]针对含巯基的β-内酰胺酶,合成了一系列硼酸类酶抑制剂.氨苄青霉素的手性类似物12针对SHVK234R的Ki值为2.4 nmol/L,为目前本类化合物中活性最佳的抑制剂.结构中使用较多的SHVK234R而非SHV1构建了复杂的氢键结构,所以进一步对氢键结构的研究将有助于发现新的β-内酰胺酶抑制剂.

图8 硼酸的抑制机理和针对β-内酰胺酶的头孢菌素机理Fig.8 Inhibition mechanisms of boronic acid and cephalothin against β-lactamase
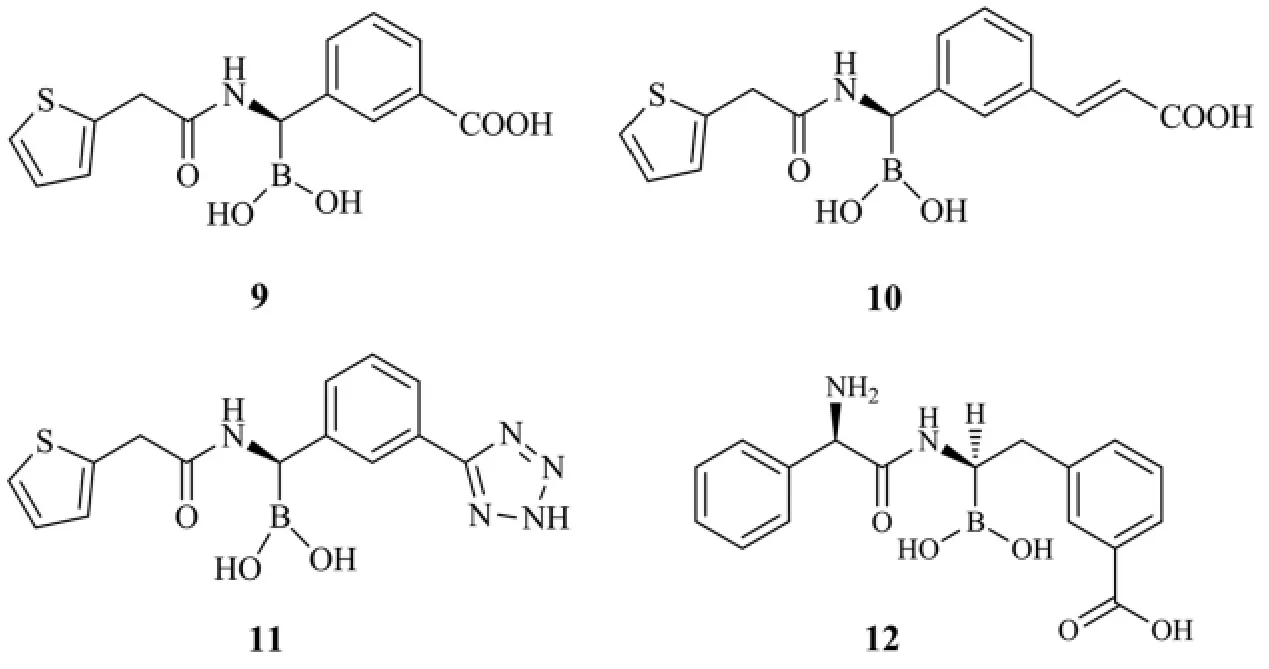
图9 化合物9,10,11和12的分子结构Fig.9 Molecular structure of compound 9,10,11 and 12
3.2.2芳香类硼酸
许多简单的芳香类硼酸表现出高效的AmpC-β-内酰胺酶抑制性( Ki值保持在微摩尔范围内),比如间硝基苯硼酸和对羧基苯硼酸.科学家设计合成了一系列3-氨基苯硼酸类衍生物,特别是噻吩磺胺类硼酸衍生物13拥有良好的β-内酰胺酶抑制活性( Ki=83 nmol/L)[30].TONDI等[31]根据AmpC-13的共晶结构,在苯硼酸的对位引入羧基发现了具有更高活性的AmpC-β-内酰胺酶抑制剂14和15.这两个化合物( Ki<30 nmol/L)的AmpC-β-内酰胺酶抑制性比化合物13多3~4倍.研究表明化合物15对位的羧基有利于与Gln120发生反应,从而产生高效抑制活性.

图10 化合物13,14和15的分子结构Fig.10 Molecular structure of compound 13,14 and 15
TONDI等[32]报道了一类广谱的硼酸类抑制剂,对A类和C类丝氨酸类β-内酰胺酶具有良好的抑制性.硼酸过渡状态的类似物能够逆转由A类和C类β-内酰胺酶产生的抑制性.苯并( b)噻吩-2-硼酸( BZB TH2B,16)为一个高效的非β-内酰胺酶的AmpC抑制剂,可以显著增强第三代头孢菌素针对中高度耐药细菌的耐药性.利用CTX-M-9,TEM-1,AmpC-β-内酰胺酶对硼酸化合物16,17,18进行效价试验,在抑制β-内酰胺酶的细菌耐药性实验中,化合物17为该类最佳的酶抑制剂(表1).
3.3中丝氨酸蛋白酶抑制剂
丙型肝炎病毒( HCV)的感染导致慢性肝疾病,比如肝硬化、肝衰竭和肝癌.作为一个临床验证的靶标,丙肝病毒NS3/4A丝氨酸蛋白酶在丙肝病毒复制中扮演重要的角色.近年来的临床研究中,科学家相继合成了一系列丙肝病毒的蛋白酶抑制剂[33-34].特拉匹韦( VX-950,Incivek和Incivo)和博赛泼维( SCH-503034,波普瑞韦)于2011年在美国批准上市.

表1 丝氨酸β-内酰胺酶BZB2THB衍生物的Ki值Table 1 The Kivalue of Serine beta lactamase BZB2THB derivatives

图11 化合物19和20的分子结构Fig.11 Molecular structure of compound 19 and 20
在2010年,纳科制药公司报道了一系列α-氨基环状硼酸衍生物,比如丙肝病毒的丝氨酸蛋白酶抑制剂.在VX-950和SCH-503034的结构上引入了不同结构的环形硼酸盐基团,得到了以五元硼酸盐19和20为代表的化合物( IC50均为0.12 μmol/L)[35].通过X射线的晶体结构研究发现,化合物20结构中丝氨酸蛋白酶的结构与先导结构SCH-503034类似,催化剂Ser-139在抑制氧硼戊环开环过程中起到至关重要的作用.
3.4 DDP-IV抑制剂
DDP -IV( OPP-IV; E.C.3.4.14.5)是一种广泛分布于哺乳动物细胞和组织中的丝氨酸蛋白酶.由于DDP-IV在肠促胰岛素GLP-1和GIP中的降解作用,使其成为一类治疗Ⅱ型糖尿病的新靶点[36-37].目前,4个DPP-IV抑制剂(西他列汀、沙格列汀、维达列汀、利拉利汀)已用于Ⅱ型糖尿病的治疗.

图12 化合物21和22的分子结构Fig.12 Molecular structure of compound 21 and 22
1990年,首先报道了DPP-IV抑制剂二肽类硼酸化合物21 ( Val-boroPro)[38]和22 ( Ala-boro-Pro)[39].化合物21对DPP-IV具有良好的生物活性,但由于在斯普拉格-杜勒鼠中的毒性,科学家排除了其成为治疗糖尿病的临床候选药物的可能.化合物21的软药衍生物的研究旨在提高抗高血糖活性和降低毒性.如图13所示,体内不活跃的软药衍生物遇到DPP-IV的目标酶,导致N端二肽裂解,在生理pH条件下,化合物21活化从而抑制DPP-IV.目前,科学家正在转移性结直肠癌病人体内进行化合物21的Ⅱ期临床试验[40].

图13 DPP-IV抑制剂的前体药物策略Fig.13 Prodrug strategy of DPP-IV inhibitors
硼脯氨酸类二肽基硼酸是最早的也是最有效的DPP-IV抑制剂.WU等[40]通过利用不同基团在硼脯氨酸环的第4位进行特异性和立体选择性代替,使氨基硼酸结合到4位取代的硼脯氨酸型二肽化合物中.在该二肽类硼酸化合物中,精氨酸-( 4S) -硼羟脯氨酸23是最有效的DPP-IV,DPP8和DPP9抑制剂,相对于DPP8和DPP9,( 4S) -羟脯氨酸( 4R) -硼羟脯氨酸24对DPP-IV的选择性最强.

图14 化合物23和24的分子结构Fig.14 Molecular structure of compound 23 and 24
4 组蛋白去乙酰化酶抑制剂
组蛋白去乙酰化酶( HDACs)是一类在染色质重构、表观遗传调控的基因表达以及其他重要生物功能中扮演多个角色的一类酶.组蛋白去乙酰化酶可以去除组蛋白ε-N-乙酰基赖氨酸氨基酸上的乙酰基,可分为两类: NAD+依赖酶和Zn2+依赖酶.近年来,人们发现许多具有组蛋白去乙酰化酶抑制性的化合物,比如PXD-101[41]、曲古抑菌素A ( TSA)[42]、MS-275[43]、罗咪酯肽( FK-228)和辛二酰苯胺异羟肟酸( SAHA,伏立诺他)[44].其中,SAHA和罗咪酯肽已被批准进行治疗皮肤T细胞淋巴瘤以及丙戊酸的Ⅲ期临床试验[45].
在2009年,SUZUKI等[46]基于已提出的组蛋白去乙酰化酶抑制剂的催化机制,发现了包含α-氨基酸在内的硼酸类组蛋白去乙酰化酶抑制剂25-27.其中,S构型比R构型表现出更高的抑制活性,特别是化合物( S) -27在毫微摩尔的浓度下表现出高效的组蛋白去乙酰化酶抑制性.

图15 化合物25,26和27的分子结构和IC50值Fig.15 Molecular structure and IC50value ofcompound 25,26 and 27
5 亮氨酰-tRNA合成酶抑制剂
亮氨酰-tRNA合成酶是一类在人体内是以LARS基因编码的酶.Anacor制药公司研发了乙二酸硼酸衍生物5-氟-1,3-二氢-1-羟基-2,1-苯硼酸作为一种抗真菌剂,在甲藓的两项Ⅲ期试验中抑制活性比较理想[47].科学家利用反向遗传学的策略识别了化合物28的靶标酶.AN2690通过在酶的修饰区形成一个稳定的t-RNA-AN2690加合物,从而抑制亮氨酰tRNA合成酶.通过在这个活性修饰区内绑定tRNA达到阻止催化反转的作用,从而产生了亮氨酰tRNA的合成抑制以及蛋白合成阻塞[48].目前,MADURA等已经确认和报告了AN2690晶体结构,它们首先通过平面分子中强的O—H…O氢键形成中心对称的二聚物,然后通过分子间C—H …F和C—H…O氢键把二聚体排列成层,最后通过C—H…O的交互作用形成三维结构[49].

图16 化合物28的分子结构Fig.16 Molecular structure of compound 28
6 自毒素抑制剂
自毒素抑制剂( ATX)分泌核苷酸焦磷酸酶和磷酸二酯酶,充当了溶血磷脂酶D的作用,产生脂质介质的溶血磷脂酸( LPA)[50].自毒素抑制剂信号轴在纤维变性疾病、血管新生、慢性炎症和肿瘤进展中起着重要的作用[51].
ALBERS等[52]使用硼酸代替了HA51( 29)中的羧酸得到了自毒素抑制剂HA130,其活性提升了200倍.通过对化合物30侧链的修饰,得到了两个有效的胺类抑制剂(化合物31和化合物32).相比化合物30此两种硼酸衍生物的抑制活性并没有大幅提升,但研究证明在结构优化中硼酸基团是自毒素抑制剂的必要官能团.
7 HCV(丙型肝炎病毒)的RNA依赖性聚合酶抑制剂
硼酸基团可以增强体外抑制野生型肝炎复制子C的活性,同时抑制了HCV变异复制子的活性.通过对HCV复制子相关合成、优化、构效关系的研究,SMITH等[53]在次基因组复制系统中发现了丙肝病毒的RNA依赖性聚合酶( NS5B)抑制剂.
硼酸是羟基蛋白酶可逆性抑制的理想官能团,科学家发现了一系列有效的NS5B聚合酶抑制剂.针对关键阻力突变体的效能,科学家通过结构优化得到了临床候选化合物33.
2011年,化合物33在丙肝病毒感染受试者体内进行了临床试验.在复制周期的起始阶段,临床候选化合物33有效的抑制了RNA聚合酶的活性,离解半衰期结果大于40 h,对孤立GT1b 316N蛋白质的结合动力学性质较弱.SMITH等[53]报道了一个完整的病毒(包括化合物33在内的的一系列特异性抑制作用的复制子)以及化合物33在小白鼠、大白鼠、狗、猴子和人体中的药代动力学特征.化合物33与安慰剂(-0.09 log10copies/mL)在丙型肝炎病毒感染者体内24 h后计量比较,在单剂量420 mg(-1.33 log10copies/mL)下血清丙肝病毒核糖核酸显著减少.
8 金黄色葡萄球菌NorA(诺拉)外排泵的硼酸类抑制剂
射流泵的超表达是一种把抗菌物质挤压到细菌细胞外的细菌耐药性机制.通过抑制这种泵可以恢复现有抗生素效力.通过影响金黄色葡萄球菌的诺拉外排泵可以抑制一系列基质(比如亲水性氟喹诺酮),从而产生多药耐药性作用.科学家[54]对大约150个杂环硼酸化合物的金黄色葡萄球菌的敏感性和抵抗性进行评估.虽然24个苗头化合物单独测试时并不活跃,但当浓度范围从0.5 mg/L增加到8 mg/L时可以显著增强环丙沙星抑制金黄色葡萄球菌的活性,而超表达的诺拉无硼类似物并没有此活性,说明硼酸官能团对于射流泵的超表达是至关重要的.

图17 化合物29,30,31和32的分子结构和IC50值Fig.17 Molecular structure and IC50value of compound 29,30,31 and 32

图18 化合物33的分子结构Fig.18 Molecular structure of compound 33
6-苄氧基吡啶-3-硼酸34和4-苄氧基苯硼酸35是目前最有前景的NorA抑制剂.当MMC4值分别在1和0.5 mg/L时可以显著增强环丙沙星的活性,但内在抗菌活性并不明显,同时降低了对人类细胞系的细胞毒性.科学家正在通过对化合物5f进行药学修饰以得到更好的诺拉抑制剂.

图19 化合物34和35的分子结构Fig.19 Molecular structure of compound 34 and 35
9 CXCR1和CXCR2的非竞争性硼酸抑制剂
G蛋白偶联趋化因子受体CXCR1和CXCR2在炎性疾病和致癌作用中扮演着关键的角色.炎症发生时,通过绑定的趋化因子CXCL1 ( CXCR1)和CXCL8 ( CXCR1和CXCR2)以激活和补充多形核白细胞( PMNs).CXCL1刺激人体中性粒细胞,导致Ca2+外流,实验验证了一系列的新颖的S-6-巯基-N-苯基-烟酰胺的效应[55],发现了一个强有效的非竞争性硼酸CXCR1/2拮抗剂: 2-[5-( 4-氟苯基氨基甲酰)吡啶-2-磺酸基甲基]苯硼酸36.

图20 化合物36的分子结构Fig.20 Molecular structure of compound 36
在人体中性粒细胞内,化合物36抑制由CXCL1诱导的Ca2+外流( IC50= 38 nmol/L),但是对由C5a、fMLF或PAF诱导下Ca2+的外流没有影响.在重组的HEK293细胞内CXCR2稳定表达,SR-517拮抗由CXCL8诱导产生的(35s) GTPγS结合( IC50= 60 nmol/L)以及ERK1 /2的磷化.硼酸化合物36是一个非竞争性抑制剂,无法影响 [125I]-CXCL8与CXCR2细胞膜的结合.硼酸化合物36 ( 0.2 mg/kg iv)可显著地抑制小鼠体内的炎症.硼酸化合物33是第一个报道的硼酸类趋化因子拮抗剂以及拮抗CXCR1 /2的特异性药效团.
10 其他硼酸类化合物
近年来报道了许多可以作为特异性配体的硼酸化合物,比如类固醇硫酸酯酶抑制剂、酰胺水解酶抑制剂、一氧化氮合成酶( NOS)抑制剂、表皮生长因子受体( EGFR)酪氨酸激酶激素抑制剂、敏感性脂解酶( HSL)抑制剂、人类酰基蛋白硫酯酶( APT)抑制剂等.在表2中对这些代表化合物的结构和活性进行了总结.

表2 其他已报告硼酸类的酶抑制剂Table 2 Other boronic acid-based enzyme inhibitors have been reported

续表2
11 结论
近年来,硼酸类化合物作为酶抑制剂和特异性配体在新药发现方面取得了不凡的成就.2003年硼替佐米获得美国FDA批准上市后,研究人员相继合成和报道了一系列具有高效抑制活性以及低副作用的硼酸类化合物,其中部分已经进入临床试验.利用最新研究技术和方法工艺推进硼酸类化合物的深入研究已成为一个热门的研究方向.硼酸化合物在新药发现中前景广阔,我们相信未来必将会出现一批特异性强、安全高效的硼酸类药物.
参考文献:
[1]ZHENG N,LI H,SUN G,et al.Synthesis and properties of T-shaped organic conjugates based on 3,6-diarylpyridazine-fused tetrathiafulvalene [J].Org Biomol Chem,2013,11( 31) : 5100-5108.
[2]HAMILTON A E,BUXTON A M,PEEPLES C,et al.An operationally simple aqueous Suzuki-Miyaura cross-coupling reaction for an undergraduate organic chemistry laboratory [J].J Chem Educ,2013,90( 11) : 1509-1513.[3]吴忠玉,方浩,徐文方.基于有机硼酸的葡萄糖荧光传感器的研究进展[J].有机化学,2007,27( 7) : 830 -836.
[4]WU Z,LI M,FANG H,et al.A new boronic acid based fluorescent reporter for catechol [J ].Bioorg Med Chem Lett,2012,22( 23) : 7179-7182.
[5 ]LIENHARD G E,KOEHLER K A.2-phenylethaneboronic acid,a possible transition-state analog for chymotrypsin [J].Biochemistry-US,1971,10( 13) : 2477-2483.[6]MATTHEWS D A,ALDEN R A,BIRKTOFT J J,et al.X-ray crystallographic study of boronic acid adducts with subtilisin BPN'( Novo).A model for the catalytic transition state [J].J Biol Chem,1975,250( 18) : 7120-7126.
[7 ]EIDAM O,ROMAGNOLI C,CASELLI E,et al.Design,synthesis,crystal structures and antimicrobial activity of sulfonamide boronic acids as β-lactamase inhibitors [J].J Med Chem,2010,53( 21) : 7852-7863.
[8]KNOTT K,FISHOVITZ J,THORPE S B,et al.N-Terminal peptidic boronic acids selectively inhibit human ClpXP [J ].Org Biomol Chem,2010,8( 15) : 3451-3456.
[9 ]POURNARAS S,POULOU A,TSAKRIS A.Inhibitorbased methods for the detection of KPC carbapenemase-producing Enterobacteriaceae in clinical practice by using boronic acid compounds [J ].J Antimicrob Chemoth,2010,65 ( 7) : 1319-1321.
[10 ]BRETT L K,WILLIAMS M E.Current and emerging therapies in mantle cell lymphoma [J ].Curr Treat Option On,2013,14( 2) : 198-211.
[11]DICK L R,FLEMING P E.Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy [J].Drug Discov Today,2010,15( 5) : 243-249.
[12 ]SPRINGSTEEN G,WANG B.A detailed examination of boronic acid-diol complexation [J ].Tetrahedron,2002,58( 26) : 5291-5300.
[13]WISKUR S L,LAVIGNE J J,AIT-HADDOU H,et al.pKavalues and geometries of secondary and tertiary amines complexed to boronic acids-implications for sensor design [J].Org Lett,2001,3( 9) : 1311-1314.
[14]DONOHUE T M,CEDERBAUM A I,FRENCH S W,et al.Role of the proteasome in ethanol-induced liver pathology [J].Alcohol Clin Exp Res,2007,31( 9) : 1446 -1459.
[15]ESHAGHIAN S,BER ENSON J R.Multiple myeloma: improved outcomes with new therapeutic approaches [J].Curr Opin Support Palliat Care,2012,6( 3) : 330-336.
[16]KRAMER H B,NICHOLSON B,KESSLER B M,et al.Detection of ubiquitin-proteasome enzymatic activities in cells: Application of activity-based probes to inhibitor development [J ].Biochim Biophys Acta,2012,1823 ( 11) : 2029-2037.
[17]O'CONNOR O A,CZUCZMAN M S.Novel approaches for the treatment of NHL: Proteasome inhibition and immune modulation [J ].Leukemia Lymphoma,2008,49 ( S1) : 59-66.
[18]BERKOWITZ A,WALKER S.Bortezomib-induced peripheral neuropathy in patients with multiple myeloma [J].Clin J Oncol Nurs,2012,16( 1) : 86-89.
[19]CORSO A,MANGIACAVALLI S,VARETTONI M,et al.Bortezomib-induced peripheral neuropathy in multiple myeloma: a comparison between previously treated and untreated patients [J].Leukemia Res,2010,34( 4) : 471 -474.
[20]WATANABE T,MOMOSE I,ABE M,et al.Synthesis of boronic acid derivatives of tyropeptin: proteasome inhibitors [J].Bioorg Med Chem Lett,2009,19( 8) : 2343-2345.
[21]ZHU Y,YAO S,XU B,et al.Design,synthesis and biological evaluation of tripeptide boronic acid proteasome inhibitors [J ].Bioorgan Med Chem,2009,17( 19) : 6851-6861.
[22]MADALA P K,TYNDALL J D A,NALL T,et al.Update 1 of: Proteases universally recognize beta strands in their active sites [J].Chem Rev,2011,110( 6) : PR1-PR31.
[23]DONOHUE T M,CEDERBAUM A I,FRENCH S W,et al.Role of the proteasome in ethanol-induced liver pathology [J].Alcohol Clin Exp Res,2007,31( 9) : 1446 -1459.
[24]LAZAROVA T I,JIN L,RYNKIEWICZ M,et al.Synthesis and in vitro biological evaluation of aryl boronic acids as potential inhibitors of factor Xia [J ].Bioorg Med Chem Lett,2006,16( 19) : 5022-5027.
[25 ]NAKAYAMA S,TRIBUDDHARAT C,PROMBHUL S,et al.Molecular analyses of TEM genes and their corresponding penicillinase-producing Neisseria gonorrhoeae isolates in Bangkok,Thailand [J].Antimicrob Agents Ch,2012,56( 2) : 916-920.
[26]OHNISHI M,ONO E,SHIMUTA K,et al.Identification of TEM-135 β-lactamase in penicillinase-producing Neisseria gonorrhoeae strains in Japan [J ].Antimicrob Agents Ch,2010,54( 7) : 3021-3023.
[27]LIU J,TSAI M D.DNA polymerase β: pre-steady-state kinetic analyses of dATPαS stereoselectivity and alteration of the stereoselectivity by various metal ions and by site-directed mutagenesis [J ].Biochemistry-US,2001,40 ( 30) : 9014-9022.
[28]MORANDI S,MORANDI F,CASELLI E,et al.Structure-based optimization of cephalothin-analogue boronic acids as β-lactamase inhibitors [J ].Bioorgan Med Chem,2008,16( 3) : 1195-1205.
[29]WINKLER M L,RODKEY E A,TARACILA M A,et al.Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable ( SHV)β-lactamase [J ].J Med Chem,2013,56( 3) : 1084-1097.
[30]TONDI D,POWERS R A,CASELLI E,et al.Structure-based design and in-parallel synthesis of inhibitors of AmpC β-lactamase [J].Chem Biol,2001,8( 6) : 593-610.
[31]TONDI D,CALòS,SHOICHET B K,et al.Structural study of phenyl boronic acid derivatives as AmpC β-lactamase inhibitors [J].Bioorg Med Chem Lett,2010,20 ( 11) : 3416-3419.
[32]TONDI D,VENTURELLI A,BONNET R,et al.Targeting class a and c serine β-lactamases with a broad-spectrum boronic acid derivative [J ].J Med Chem,2014,57( 12) : 5449-5458.
[33]MCCAULEY J A,MCINTYRE C J,RUDD M T,et al.Discovery of vaniprevir ( MK-7009),a macrocyclic hepatitis C virus NS3/4a protease inhibitor [J].J Med Chem,2010,53( 6) : 2443-2463.
[34 ]TORBEEV V Y,MANDAL K,TERECHKO V A,et al.Crystal structure of chemically synthesized HIV-1 protease and a ketomethylene isostere inhibitor based on the p2/ NC cleavage site [J].Bioorg Med Chem Lett,2008,18 ( 16) : 4554-4557.
[35]LI X,ZHANG Y K,LIU Y,et al.Synthesis and evaluation of novel α-amino cyclic boronates as inhibitors of HCV NS3 protease [J].Bioorg Med Chem Lett,2010,20( 12) : 3550-3556.
[36]KIEFFER T J,FRANCIS HABENER J.The glucagonlike peptides [J ].Endocr Rev,1999,20( 6) : 876-913.
[37]REIMER M K,HOLST J J,AHRÉN B.Long-term inhibition of dipeptidyl peptidase IV improves glucose tolerance and preserves islet function in mice [J].Eur J Endocrinol,2002,146( 5) : 717-727.
[38]NARRA K,MULLINS S R,LEE H O,et al.Phase II trial of single agent Val-boroPro ( Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer [J ].Cancer Biol Ther,2007,6( 11) : 1691-1699.
[39]REINHOLD D,BITON A,GOIHL A,et al.Dual inhibition of dipeptidyl peptidase IV and aminopeptidase N suppresses inflammatory immune responses [J ].Ann Ny Acad Sci,2007,1110( 1) : 402-409.
[40]WU W,LIU Y,MILO L J,et al.4-Substituted boroproline dipeptides: Synthesis,characterization,and dipeptidyl peptidase IV,8,and 9 activities [J].Bioorg Med Chem Lett,2012,22( 17) : 5536-5540.
[41 ]RAMALINGAM S S,BELANI C P,RUEL C,et al.Phase II study of belinostat ( PXD101),a histone deacetylase inhibitor,for second line therapy of advanced malignant pleural mesothelioma [J].J Thorac Oncol,2009,4 ( 1) : 97.
[42]HAN S,LU J,ZHANG Y,et al.HDAC inhibitors TSA and sodium butyrate enhanced the human IL-5 expression by altering histone acetylation status at its promoter region [J].Immunol Lett,2007,108( 2) : 143-150.
[43 ]BARADARI V,HOPFNER M,HUETHER A,et al.Histone deacetylase inhibitor MS-275 alone or combined with bortezomib or sorafenib exhibits strong antiproliferative action in human cholangiocarcinoma cells [J ].World J Gastroentero,2007,13( 33) : 4458.
[44]MIELCAREK M,BENN C L,FRANKLIN S A,et al.SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease [J].PLoS One,2011,6( 11) : e27746.
[45 ]GUAN P,FANG H.Clinical development of histone deacetylase inhibitor Romidepsin [J ].Drug Discov T-her,2010,4( 6) : 388-391.
[46]SUZUKI N,SUZUKI T,OTA Y,et al.Design,synthesis,and biological activity of boronic acid-based histone deacetylase inhibitors [J].J Med Chem,2009,52( 9) : 2909-2922.
[47 ]HUI X,BAKER S J,WESTER R C,et al.In vitro penetration of a novel oxaborole antifungal ( AN2690) into the human nail plate [J].J Pharm Sci,2007,96( 10) : 2622-2631.
[48]ROCK F L,MAO W,YAREMCHUK A,et al.An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site [J ].Science,2007,316( 5832) : 1759-1761.
[49]MADURA I D,ADAMCZYK-WOZNIAK A,JAKUBCZYK M,et al.5-Fluoro-1,3-dihydro-2,1-benzoxaborol-1-ol [J].Acta Crystallogr E,2011,67( 2) : 0414-0415.[50]STRACKE M L,KRUTZSCH H C,UNSWORTH E J,et al.Identification,purification,and partial sequence analysis of autotaxin,a novel motility-stimulating protein [J].J Biol Chem,1992,267( 4) : 2524-2529.
[51]VAN MEETEREN L A,MOOLENAAR W H.Regulation and biological activities of the autotaxin-LPA axis [J].Prog Lipid Res,2007,46( 2) : 145-160.
[52]ALBERS H M H G,DONG A,VAN MEETEREN L A,et al.Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation [J ].P Natl A Sci,2010,107( 16) : 7257-7262.
[53]MAYNARD A,CROSBY R M,ELLIS B,et al.Dis-covery of a potent boronic acid derived inhibitor of the HCV RNA-dependent RNA polymerase [J ].J Med Chem,2013,57( 5) : 1902-1913.
[54]FONTAINE F,HEQUET A,VOISIN-CHIRET A S,et al.First identification of boronic species as novel potential inhibitors of the Staphylococcus aureus NorA efflux pump [J].J Med Chem,2014,57( 6) : 2536-2548.
[55]MAEDA D Y,PECK A M,SCHULER A D,et al.Discovery of 2-[5-( 4-Fluorophenylcarbamoyl) pyridin-2-ylsulfanylmethyl]phenylboronic Acid ( SX-517) : Noncompetitive Boronic Acid Antagonist of CXCR1 and CXCR2 [J].J Med Chem,2014,57( 20) : 8378-8397.
[责任编辑:任铁钢]
Research progress of boronic acid compounds in drug discovery
WANG Kai1,2,3,WU Zhongyu1,2,3,YAO Qingqiang1,2,3*,SUN Jingyong1,2,3
( 1.Institute of Materia Medica,Shandong Academy of Medical Sciences,Jinan 250062,Shandong,China; 2.School of Medicine and Life Sciences,University of Jinan-Shandong Academy of Medical Sciences,Jinan 250062,Shandong,China; 3.Key Lab.of Rare and Uncommon Diseases of Shandong Province,Jinan 250062,Shandong,China)
Abstract:Boronic acid compounds are widely applied to organic synthesis,fluorescent sensing,drug discovery,etc.Especially since bortezomib was approved by FDA,scientists have devoted great interest in boronic aicd-based drug discovery.This paper reviewed the latest progress of boronic acid compounds in new drug discovery including enzyme inhibitors and specific ligands.
Keywords:boronic acid compound; enzyme inhibitor; bortezomib; drug discovery
作者简介:王凯( 1989-),男,硕士生,专业方向为药物化学.*通讯联系人,E-mail: yao_imm@ 163.com.
基金项目:山东省自然科学基金资助项目( ZR2014YL035),山东省医学科学院院级科技计划项目( 2013-34),2014年度留学人员科技活动项目择优资助.
收稿日期:2014-08-17.
中图分类号:O627
文献标志码:A
文章编号:1008-1011( 2016) 01-0021-13
