超滤管净化/高效液相色谱-串联质谱法测定动物源食品中喹诺酮类药物残留
2016-02-23鞠玲燕宋晓华徐成钢杨丽君
鞠玲燕,宋晓华,谷 婕,徐成钢,杨丽君
( 威海出入境检验检疫局,山东 威海 264200)
超滤管净化/高效液相色谱-串联质谱法测定动物源食品中喹诺酮类药物残留
鞠玲燕,宋晓华,谷婕,徐成钢,杨丽君*
( 威海出入境检验检疫局,山东威海264200)
喹诺酮类(Quinolones,QNs)药物是一类抗菌作用强、抗菌谱广的人工合成抗菌药,广泛应用于临床诊断、动物疾病预防及促生长。QNs在动物源食品中的过量或不当使用会对食用者产生潜在“三致”(致癌、致畸、致突变)作用,诱导致病菌产生耐药性,从而威胁人类健康[1]。因此,动物体内的QNs残留问题备受关注,许多国家和组织都限制其使用并制订出相应的最高残留限量(MRLs):美国禁止在食用动物养殖中使用氟喹诺酮类药物(Fluoroquinolones, FQNs);日本批准使用的QNs仅有恩诺沙星、二氟沙星、奥比沙星、达氟沙星、马波沙星、氧氟沙星和恶喹酸;欧盟规定动物肌肉、肝脏和肾脏中达氟沙星、二氟沙星、恩诺沙星(环丙沙星与恩诺沙星量之和)、麻保沙星、沙拉沙星等的MRLs为0.01~1.9 mg;我国于2002年规定了环丙沙星、单诺沙星、恩诺沙星、沙拉沙星、二氟沙星、恶喹酸和氟甲喹等7种QNs药物在动物肌肉组织中的最高残留限量为10~500 μg/kg。因此,对动物源食品中QNs残留量的检测尤为重要。
目前QNs的检测方法主要有高效液相色谱法(HPLC)[2-3]、高效液相色谱-串联质谱法(HPLC-MS/MS)[4-5]、酶联免疫法(ELISA)[6]等。其中HPLC-MS/MS法分析速度快、灵敏度高、选择性和特异性好,是目前药物残留方面应用最普遍的检测技术[7]。样品前处理技术对于药物的检测至关重要[8]。QNs样品前处理的常用方法有液液萃取(LLE)和固相萃取(SPE);LLE是净化QNs的基本方法,但存在操作费时、消耗高纯溶剂多以及样品易乳化等不足[9-10];SPE消耗溶剂少且不产生乳化现象,是QNs净化最常用的方法[11-13],但目前商品化的固相萃取柱一般只能一次性使用,成本较高。另外还有分子印迹固相萃取、基质固相分散萃取和超临界流体萃取等较新的方法也被应用于QNs残留分析,但均处于研究阶段,技术尚不成熟[14-15]。
超滤(Ultrafiltration,UF)是一种加压膜分离技术,其膜孔径介于微滤和反渗透之间,通过膜表面的微孔结构对物质进行选择性分离。即在一定压力下,使小分子物质穿过一定孔径的特制薄膜,而大分子物质不能透过,从而将小分子与生物大分子分开的一种分离纯化技术[16-18]。目前超滤技术较多应用于水处理工程,将其与HPLC-MS/MS结合应用于兽残检测的研究较少。本研究尝试利用实用和快捷的超滤管净化样品,采用高效液相色谱-四极杆串联质谱同时测定动物源食品中的10种喹诺酮类药物残留量,有效地消除了基质效应,方法灵敏度高、重复性好,适合于动物源食品中多种喹诺酮类药物残留的快速确证和定量测定。
1实验部分
1.1仪器、试剂与材料
Agilent 1200高效液相色谱仪(美国Agilent公司);API 4000串联四极杆质谱仪(美国AB公司);配有电喷雾离子源(ESI);CR22GⅡ高速冷冻离心机(日立公司);氮吹浓缩仪(美国Caliper公司);MS3 旋涡混合器(IKA公司)。
乙腈(色谱纯,美国默克公司);甲酸、乙酸(色谱纯,CNW公司);无水硫酸钠(分析纯,天津市大茂化学试剂厂):650 ℃灼烧4 h,置于干燥器中备用;1%乙酸酸化乙腈:99 mL乙腈中加入1 mL乙酸,混匀。标准品:恩诺沙星(ENR)、环丙沙星(CIP)、恶喹酸(OXO)、沙拉沙星(SAR)、双氟沙星(DIF)、氟甲喹(FLU)、萘啶酸(NDL)、氧氟沙星(OFL)、诺氟沙星(NOR)、丹诺沙星(DAN)均购自Dr.Ehrenstorfer,纯度≥97.2%。
超滤管:体积为50 mL,截留分子量(MWCO):选用3 kD和10 kD,购自美国Millipore公司。新超滤管使用前加入超纯水,水量完全过膜,冰浴或冰箱中预冷几分钟,将水倒出,然后加入前处理液。
1.2标准溶液的制备
用乙腈溶解标准品,配成浓度为100 mg/L的单标储备液,于-18 ℃下储存备用。使用时根据需要配成适宜浓度的混合标准中间液和标准工作液。
1.3液相色谱条件
色谱柱:Waters Xbridge C18(3.5 μm×2.1 mm×150 mm);进样量:20 μL;流动相:A为0.1%甲酸溶液;B为乙腈;流速:0.2 mL/min;梯度洗脱条件:0~1.0 min,80%A;1.0~6.0 min,80%~40%A;6.0~10.0 min,40%~20%A;10.0~14.0 min,20%A;14.0~14.1 min,20%~80%A;14.1~20.0 min,80%A。
1.4质谱条件
电喷雾电离源正离子扫描(ESI+);多反应监测模式(MRM);电喷雾电压:5.0 kV;离子源温度:500 ℃;雾化气压力(GS1):70 psi;辅助气流速(GS2):55 psi;气帘气压力(CUR):20 psi;碰撞室入口电压(EP):10 V;碰撞室出口电压(CXP):10 V。监测离子对、去簇电压(DP)、碰撞能量(CE)如表1所示。
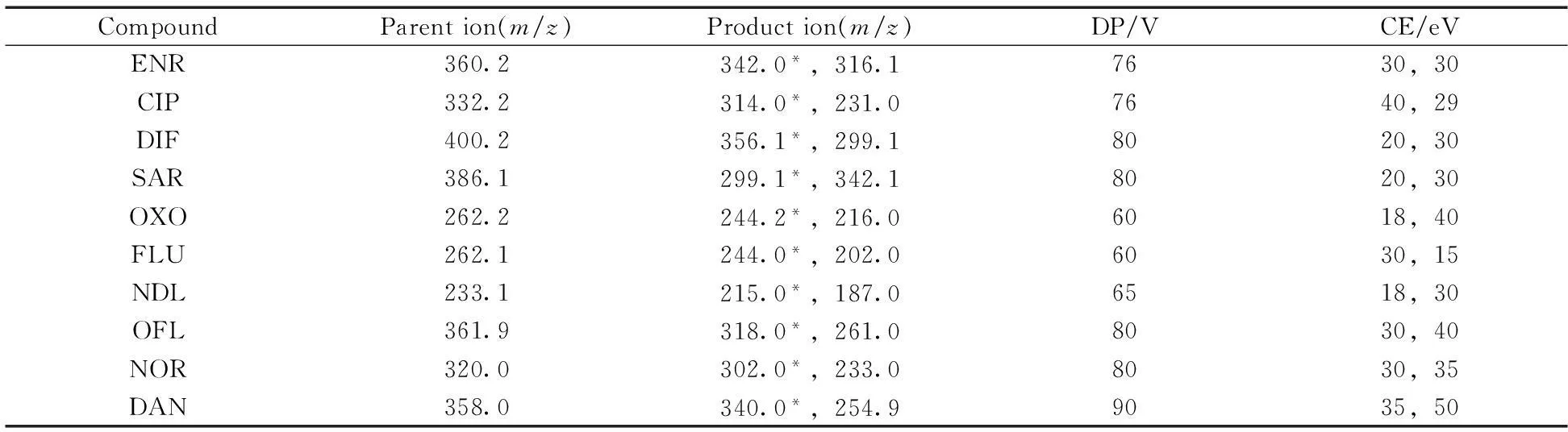
表1 10种喹诺酮的优化质谱分析参数
*quantitative ion
1.5样品前处理
准确称取5.00 g粉碎好的样品,加入20 mL 1%乙酸酸化乙腈,混匀,超声20 min,加入5 g无水硫酸钠,混匀,以8 000 r/min离心5 min,取12 mL左右上清液沿管壁慢慢加入超滤管中。离心机预冷至4 ℃,5 000 r/min离心10 min,准确移取离心出的液体10 mL用氮气流吹干,以流动相定容至1.0 mL,供HPLC-MS/MS测定分析。
2结果与讨论
2.1样品前处理条件的比较
2.1.1超滤膜分子量的选择通常情况下选择合适的超滤管,主要考虑MWCO和浓缩体积,MWCO不应大于目的蛋白分子量的1/3,若待测蛋白的分子量为10 kD左右,则可以用截留分子量3 kD的超滤管。而本实验是需要离心透析出的液体,因此在选用超滤管时不但要考虑能否透析出目标化合物,还要考虑被截留的大分子物质在离心时是否会损坏超滤膜。10种喹诺酮类药物的分子量均在200~400之间,根据现有的50 mL超滤管的型号规格(3,10,30,50,100 kD),选用膜MWCO为3 kD和10 kD的超滤管进行试验。
先加入10 mL 1%乙酸酸化乙腈离心10 min,3,10 kD超滤管均能全部滤出;选用猪肉和猪肝样品,按“1.5”方法进行样品处理后,取10 mL上清液加入超滤管中,5 000 r/min离心10 min,3,10 kD超滤膜均能够离心彻底,且未见破损。为了更好地截留生物大分子物质,达到最佳净化效果,本实验选用截留分子量最小的3 kD超滤管。
2.1.2离心条件的选择超滤管推荐的离心速率约为5 000 r/min。在空白样品中添加10种喹诺酮类药物混标(加标水平10.0 μg/kg),加入20 mL 1%乙酸酸化乙腈,混匀,超声20 min,加烘干的无水硫酸钠5 g混匀,8 000 r/min离心5 min,取10 mL上清液加入超滤管中,分别对不同离心速率(3 000,4 000,5 000,6 000 r/min)和离心时间(5,10,15,20 min)进行比较,上机检测后计算10种目标物的回收率。实验结果显示,离心速率对回收率无明显影响,与离心出的液体快慢有关,即离心速率越大,达到相同净化效果所需的离心时间越短,但离心速率过快,会损坏超滤膜,故在实际使用中采用 5 000 r/min离心10 min,其离心出的液体量满足实验要求且滤膜无破损。
2.1.3超滤管的净化效果在空白牛肉样品中添加10种喹诺酮药物混标(加标水平为5.0 μg/kg),加入20 mL 1%乙酸酸化乙腈提取后,取10 mL上清液加入超滤管中,进行净化处理,离心液经氮吹后复溶,上机检测;另取10 mL上清液直接氮吹复溶后检测。经超滤管净化处理和未处理的牛肉样品的总离子流图见图1,由图可以看出,经超滤管净化处理的样品干扰减少,降低了基质效应,达到了净化目的。
2.2液相色谱条件的选择
对比了以乙腈和0.1%甲酸、甲醇和0.1%甲酸、乙腈和水、甲醇和水作为流动相时对待测物质色谱分离的影响。结果显示,流动相中加入0.1%甲酸可以提高待测物质的信号强度;采用甲醇和乙腈为流动相时,信号强度的差别不大,但采用甲醇时的基线噪音较大,因此选用乙腈作流动相。为了达到满意的峰形和分离效果,采用乙腈-0.1%甲酸溶液作为流动相进行梯度洗脱。在色谱柱的选择中,比较了Agilent Zorbax SB-C18,XDB-C18和Waters Xbridge C183种色谱柱的分离效果,实验最终选用分离效果最佳的Waters Xbridge C18色谱柱进行分离。
2.3质谱分析条件的优化
选择ESI+模式,采用针泵进样方式分别对分析物进行质谱条件优化,每种分析物选取丰度较高的3~4个碎片离子作为子离子,配制不同种类空白样品基质标准溶液,剔除易受基质干扰的子离子。在MRM模式下优化各种质谱条件,最终选定的特征离子及优化的质谱分析参数见表2。10种喹诺酮添加水平为5.0 μg/kg的牛肉样品的MRM谱图见图2。
2.4方法学验证
2.4.1线性范围、检出限与定量下限按“1.5”方法制备不含待测组分的空白基质溶液,配制浓度分别为5.0,10.0,25.0,50.0,100.0 μg/kg的系列基质标准工作溶液,上机测定,以峰面积(y)对相应的喹诺酮浓度(x,μg/kg)绘制标准曲线。结果表明,在5.0~100 μg/kg(相当于样品)浓度范围内,10种喹诺酮的标准校正曲线线性关系良好,相关系数(r)均大于0.99。检出限(LOD)和定量下限(LOQ)采用向空白样品中逐级降低加标浓度的方法来确定。以3倍信噪比(S/N=3)对应的目标物浓度作为检出限,以S/N=10对应的目标物浓度作为定量下限。10种喹诺酮类药物的LOD为0.15~0.75 μg/kg,LOQ为0.49~2.59 μg/kg(见表2)。
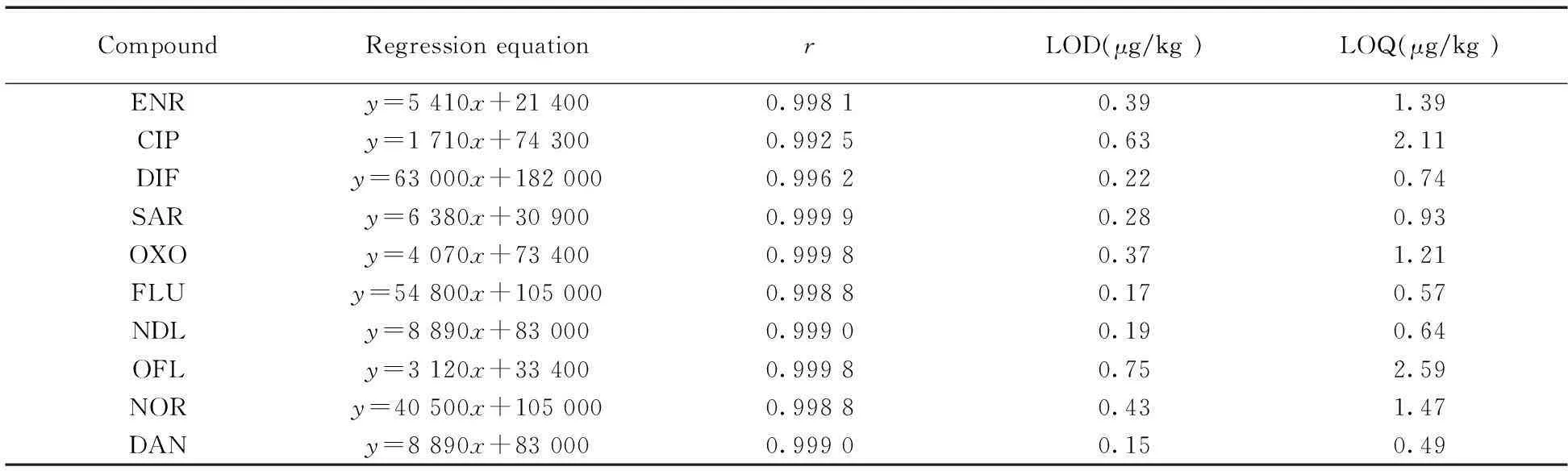
表2 10种喹诺酮的线性回归方程、相关系数、检出限与定量下限
2.4.2回收率与精密度选用不同基质(猪肝、牛肉、鱼肉)的空白样品,分别添加5.0,10,50 μg/kg 3个不同浓度水平的混合标准溶液,按“1.5”方法进行前处理,每个水平重复测定6次,计算其回收率和相对标准偏差(RSD),结果见表3。测得平均回收率为71.4%~85.9%,RSD为3.9%~10.7%。该方法具有较好的回收率和重复性,可以满足动物源性食品中10种喹诺酮类药物残留的日常检测要求。
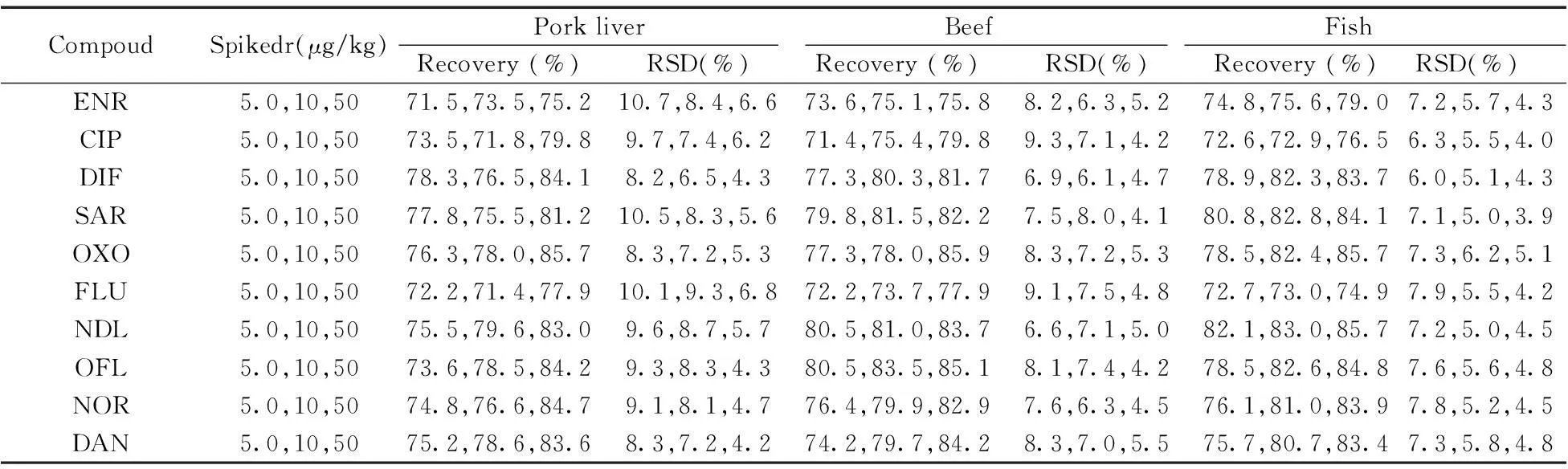
表3 猪肝、牛肉和鱼肉中10种喹诺酮的平均回收率和相对标准偏差(n=6)
2.5实际样品的测定
从市面上购得猪肝、牛肉、鱼肉等10份以及本实验室保留的阳性样品3份,分别采用本方法和《GB/T 21312-2007 动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法》进行检测,10份市售样品皆未检出10种喹诺酮药物残留,3份阳性样品中分别检出恩诺沙星、环丙沙星和诺氟沙星,将对本方法阳性样品的测定结果与国家标准方法的测定结果进行对比,测定值的相对偏差在±10%以内(见表4),说明本方法的检测结果准确可靠。
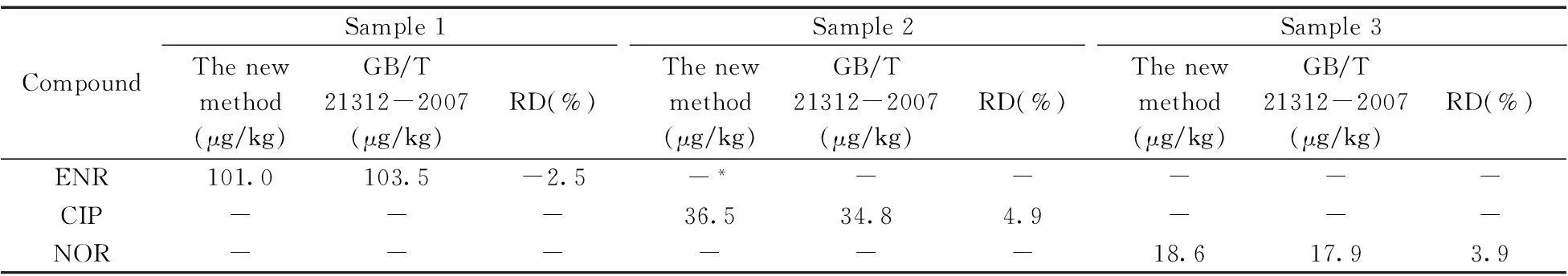
表4 阳性样品中10种喹诺酮的检测数据和相对偏差
*no detected
3结论
本研究通过优化超滤管体积、截留分子量、离心条件等参数,建立了超滤管净化/高效液相色谱-串联质谱法同时测定动物源食品中10种喹诺酮类药物残留的新方法。该方法采用超滤管净化,净化效果理想,能够实现大分子物质与残留目标物的有效分离。与传统的液液萃取(LLE)和固相萃取(SPE)相比,超滤管净化技术简便快捷、无需使用大量的有机溶剂,超滤透过的物质仅与分子量有关,与物质的其它性质无关,可去除任何样品中的生物大分子物质(如蛋白质等),有效降低样品干扰,在复杂基质中的应用效果更明显。本方法可扩展到其它兽药残留的检测,为改进动物源食品中兽药残留检测的前处理方法提供了新的契机。
参考文献:
[1]Li P P,Guo Y M,Chen X C,Zhang X J,Mei G M,Long J.FoodSci.(李佩佩,郭远明,陈雪昌,张晓军,梅光明,龙举.食品科学),2013,34(3):303-307.
[2]Lin B Y.Chin.J.Chromatogr.(林保银.色谱),2009,27(2):206-210.
[3]Li C,Jiang H Y,Wu Y L,Wang Z H,Zhao S J,Li J C,Shi Y X,Shen J Z.Chin.J.Anal.Chem.(李存,江海洋,吴银良,王战辉,赵思俊,李建成,石玉祥,沈建忠.分析化学),2009,37(8):1102-1106.
[4]Luo H T,Huang X L,Wu H Q,Zhong Q L,Zhu Z X,Huang F,Lin X S,Xie M T,Ouyang G F.J.Instrum.Anal.(罗辉泰,黄晓兰,吴惠勤,钟巧莉,朱志鑫,黄芳,林晓珊,谢梦婷,欧阳钢锋.分析测试学报),2015,34(9):979-985.
[5]Zhang Y H,Jin Y.J.Instrum.Anal.(张艳海,金燕.分析测试学报),2014,33(10):1148-1153.
[6]Zheng J,Huang X R,Li Y P,Li X J,Lin J.FoodSci.(郑晶,黄晓蓉,李耀平,李小晶,林杰.食品科学),2004,25(10):247-250.
[7]GB/T 21312-2007.Analysis of Fourteen Quinolones in Food of Animal Origin by High Performance Liquid Chromatography Tandem Mass Spectrometry.National Standards of the People’s Republic of China(动物性食品中14种喹诺酮类药物残留检测方法-液相色谱-质谱/质谱法.中华人民共和国国家标准).
[8]Li W C,Ji C Y,Zhang J J,Cheng H K,Deng C,Zhong J L,Deng X B,Shen X G.J.SouthChinaAgric.Univ.(李维铖,姬长云,张静洁,程汉奎,邓超,钟佳莲,邓衔柏,沈祥广.华南农业大学学报),2014,35(5):14-18.
[9]Peng T,Yong W,An J,Chu X G,Tang Y Z,Li C J.Chin.J.Anal.Chem.(彭涛,雍炜,安娟,储晓刚,唐英章,李重九.分析化学),2006,34(9):10-14.
[10]Zhan C R,Wen Z H,Bo Y G,Zhu J X.FoodSci.(占春瑞,温志海,卜延刚,祝建新.食品科学),2005,26(10):172-176.
[11]Hermo M P,Barron D,Barbosa J.J.Chromatogr.A,2006,1104(1/2):132-139.
[12]Bailac S,Ballesteros O,Jiménez-Lozano E,Barron D,Sanz-Nebota V,Navalón A,Vilchez J L,Barbosa J.J.Chromatogr.A,2004,1029(1/2):145-151.
[13]Zhao S J,Zheng Z R,Qu Z N,Jiang H Y,Zhang S X,Shen J Z.Chin.J.Anal.Chem.(赵思俊,郑增忍,曲志娜,江海洋,张素霞,沈建忠.分析化学),2009,37(3):335-340.
[14]Qiao F X,Sun H W,Liu G Y,Liang S X.J.HebeiUniv.:Nat.Sci.Ed.(乔风霞,孙汉文,刘广宇,梁淑轩.河北大学学报:自然科学版),2008,28(6):620-624.
[15]Shen J Y,Yin H X,Shen Z H.FoodSci.Technol.(申京宇,尹花仙,沈在汉.食品科技),2007,(10):210-212.
[16]González P,Fente C A,Franco C,Vázquez B,Qtuinto E,Cepeda A.J.Chromatogr.B,1997,693:321-326.
[18]Fente C A,Vázquez B I,Franco C,Cepeda A,Gigosos P G.J.Chromatogr.B,1999,726(1/2):133-139.
摘要:建立了超滤管净化技术结合高效液相色谱-串联质谱同时测定动物源食品中10种喹诺酮类药物残留的新方法。样品经酸化乙腈提取后,采用50 mL Millipore超滤管(截留分子量3 kD)净化,离心液氮吹复溶后,经Waters Xbridge C18色谱柱进行分离,以0.1%甲酸-乙腈作为流动相进行梯度洗脱,电喷雾正离子(ESI+)模式电离,多反应监测(MRM)模式检测,外标法定量。结果表明,10种喹诺酮类药物在5.0~100 μg/kg浓度范围内线性关系良好,方法的检出限为0.15~0.75 μg/kg,定量下限为0.49~2.59 μg/kg,在5.0,10,50 μg/kg 3个加标水平下的回收率为71.4%~85.9%,相对标准偏差(n=6)为3.9%~10.7%。方法简便快速、灵敏度高、重复性好,可用于动物源食品中10种喹诺酮类药物残留的快速确证和定量分析。
关键词:超滤管;高效液相色谱-串联质谱;动物源食品;喹诺酮类药物
Determination of Quinolones Residues in Animal-originated Foodstuffs by Ultrafiltration Tube Cleaning and High Performance Liquid Chromatography-Tandem Mass SpectrometryJU Ling-yan,SONG Xiao-hua,GU Jie,XU Cheng-gang,YANG Li-jun*
(Weihai Entry-Exit Inspection and Quarantine Bureau,Weihai264200,China)
Abstract:A new method was established for the simultaneous determination of 10 quinolone(QN) residues in animal-originated foodstuffs by ultrafiltration tube cleaning with high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS).After extraction with acetonitrile (containing 1%acetic acid),the extract was purified with 50 mL Millipore ultrafiltration tube (Molecular Weight Cut-Off,MWCO:3 kD),and then the centrifugal liquid was evaporated to dryness under a stream of nitrogen,and redissolved.The analytes were separated on a Waters Xbridge C18column using 0.1%formic acid-acetonitrile as mobile phase.The identification and quantification of 10 quinolone residues were carried out under positive electrospray ionization (ESI+) in multiple reaction monitoring (MRM) mode,the quantification analysis was performed by the external standard method.The calibration curves showed good linearities in the range of 5.0-100 μg/kg for all QNs.The limits of detection for all QNs were in the range of 0.15-0.75 μg/kg,and the limits of quantitation were 0.49-2.59 μg/kg.The recoveries of these analytes varied from 71.4%to 85.9%at spiked levels of 5.0,10,50 μg/kg,with relative standard deviations of 3.9%-10.7%.This method was simple,rapid,sensitive and reproducible,and could be applied in the determination of 10 quinolone residues in animal-originated foodstuffs.
Key words:ultrafiltration tube;high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS);animal-originated foodstuffs;quinolones
中图分类号:O657.63;TQ460.72
文献标识码:A
文章编号:1004-4957(2016)01-0042-06
doi:10.3969/j.issn.1004-4957.2016.01.007
通讯作者:*杨丽君,硕士,高级工程师,研究方向:食品安全,Tel:0631-3807680,E-mail:18663136117@163.com
基金项目:国家质检总局科技计划项目(2013IK178);质检公益性行业科研专项(201310143);国家科技支撑计划项目(2012BAK08B01)
收稿日期:2015-07-01;修回日期:2015-07-31
