Pt+促进丙烷1,2-脱氢气相反应路径中结构和能量学的理论分析
2016-02-17赵改云郭玥张干兵钟欣欣胡玮夏清华
赵改云,郭玥,张干兵,钟欣欣,胡玮,夏清华
(有机化工新材料湖北省协同创新中心,有机功能分子合成与应用教育部重点实验室,
湖北大学化学化工学院,湖北 武汉 430062)
Pt+促进丙烷1,2-脱氢气相反应路径中结构和能量学的理论分析
赵改云,郭玥,张干兵,钟欣欣,胡玮,夏清华
(有机化工新材料湖北省协同创新中心,有机功能分子合成与应用教育部重点实验室,
湖北大学化学化工学院,湖北 武汉 430062)
摘要:采用密度泛函理论方法UB3LYP,结合相对论赝势,优化Pt+活化丙烷1,2-脱氢反应路径中各基元步骤涉及的反应物、中间体、过渡态和产物的几何构型,并在同一水平上计算其振动频率和能量.结果表明,整个反应路径放热195.7 kJ/mol,决速步骤是第二个C—H键的活化,能垒是48.7 kJ/mol.说明1,2脱氢反应在常温常压下特别容易进行,计算结果与实验观察的结果一致.
关键词:丙烷活化;1,2-脱氢路径;气相铂离子催化;反应机理;密度泛函理论
0引言
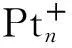
1计算方法
用密度泛函理论方法UB3LYP[13-15]对铂离子与丙烷1,2-脱氢反应路径所有驻点(反应物、中间体、过渡态和产物)的几何构型进行了全优化,并得到其能量.通过振动分析确证所优化的驻点对应局域极小或过渡态,并得到零点振动能(ZPVE)和热化学量.计算中用相对论赝势[16-17]来描述Pt的含60个内层电子的惰性原子实([Kr]4d104f14),而其5s/5p电子与5d/6s/6p价电子一起做明确处理.Pt采用经Couty和Hall改进的LANL2DZ相对论赝势基组[18],外加f极化函数[19],C和H采用标准的3-ξ基6-311G**[20].通过内禀反应坐标(IRC)[21-23]计算证实过渡态确实沿反应坐标分别连接反应物和产物.所有计算用Gaussian 09程序[24]完成.
为标定所选理论方法,先用所选方法计算了体系的有关性质,与相应实验值比较.计算得到铂的电离能为8.74 eV,与实验值9.0 eV[25]接近;Pt+-CH2解离能的计算值为493.2 kJ/mol,和实验值479.0±4.2 kJ/mol一致[10].因此,可以认为所选方法和基组对研究该反应来说是合适的.
2结果讨论
按脱氢方式的不同,Pt+使丙烷脱氢的反应可能存在多种不同路径.因Uggerud等人的同位素实验
确认按1,2-脱氢方式进行,本文中只简报对1,2-脱氢路径的理论研究结果.铂离子的基态组态为5d9,计算所得的二重态的能量最低,与洪特规则一致.计算结果显示Pt+活化丙烷脱氢反应中没有涉及自旋交叉,在下文的讨论中只涉及能量最低的二重态势能面.计算得到各个驻点(反应物、中间体、过渡态以及产物)的优化的几何结构展示在图1中;各驻点的相对能量列于表1,相应的势能曲线见图2.为方便叙述,下文中用粗体的数字表示各物种.
在丙烷1,2-脱氢路径中,Pt+分别活化一个C(α)—H和一个C(β)—H键,消除一分子氢,产物为Pt[CH2CHCH3]+.由表1可知,沿该路径整个反应放热195.7 kJ/mol(ΔG298=-196.5 kJ/mol),说明经该路径反应是热力学可能的.这与
实验发现该反应在常温常压下发生的事实一致.
如图1、2和表1所示,Pt+和丙烷形成稳定的反应络合物[PtC3H8]+(1),其中一个C(β)—H(1)键从0.107 nm被拉长至0.133 nm,而Pt-H(1)和Pt-C(β)键长值分别为0.163和0.236 nm,这表明Pt+和丙烷间形成了一个典型的agostic键.这种相互作用更加稳定了中间体1,使其能量比分离反应物的低180.1 kJ/mol.接着1经过过渡态TS1/2形成了插入中间体H-Pt+-C3H7(2).从键长值可以看到,中间体2中形成了正常的Pt—H键和Pt—C键.虚频(309.7i cm-1)表明TS1/2确实为一级鞍点.TS1/2中,C—H(1)、Pt—H(1) 和Pt—C(β)键长分别为0.141、0.159和0.231 nm,分别和1中的对应键长相近,表明TS1/2是一个典型的“早”过渡结构,以致于该活化步几乎无能垒(在不考虑零点校正的情况下,TS1/2的能量略高于中间体1).这一步放出77.6 kJ/mol的热量.

图1 Pt+活化丙烷1,2-脱氢反应路径中各驻点的优化几何(键长的单位是nm,键角的单位是
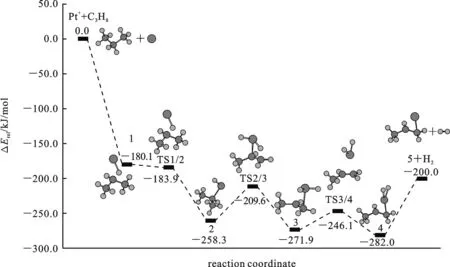
图2 用UB3LYP计算Pt+活化丙烷1,2-脱氢反应路径的二重态势能曲

表1 Pt+活化丙烷1,2-脱氢反应中各驻点的相对能量ΔErel (考虑零点能),ΔEelec (不考虑零点能),298 K下的焓ΔH298以及吉布斯自由能ΔG298(单位:kJ/mol)
然后,2中的第二个C—H键C(α)—H(2)被活化断开,同时Pt—H(2)键长由0.159 nm缩短为0.154 nm,从而形成金属二氢化物中间体3.此步经过过渡态TS2/3,翻越48.7 kJ/mol的能垒,放出12.1 kJ/mol 的热量.TS2/3的虚频939.0i cm-1确证为一个过渡态.从几何上来看,TS2/3明显地接近3的特征,表明TS2/3是典型的“迟”过渡结构.此步是整个催化过程中位垒最高的,因此成为整个过程的决速步.中间体3中两个氢配体还原消去,经过过渡态TS3/4,翻越25.8 kJ/mol的能垒,形成金属分子氢络合物4.这一步放热12.0 kJ/mol.过渡态TS3/4(266.8i cm-1)与4的结构相近,其中H—Pt—H平面和丙烯基平面是近垂直的,因此也具有“迟”过渡结构特征.从3到4,H(1)—H(2)距离缩短到0.083 nm,此距离已很接近于游离H2中的H—H键长(0.075 nm),而同时Pt—(H2)键长由0.154变长到0.179 nm,Pt—C(α)键长由0.222 nm缩短为0.219 nm.这说明4中Pt与丙烯配体作用增强,而与氢分子配体间仅有较弱的agostic相互作用.这有利于下一步H2的消去,事实上H2比较容易消去,仅需82.0 kJ/mol的能量,且生成的脱氢产物(H3CC(H)Pt CH2)+(5)与4中的相应部分结构几乎一样.中间体4在整个体系的势能面中具有最小值.
3结论
结合相对论赝势,用密度泛函理论方法UB3LYP计算了Pt+活化丙烷1,2-单分子脱氢反应路径的结构和势能面.结果显示,该反应按1,2-脱氢方式的⊿G≪0;而1,2-消除反应的决速步(第二个C—H的活化步)的能垒很小,仅为48.7 kJ/mol.表明反应沿1,2-脱氢路径,在热力学和动力学上都是有利的,与实验发现该反应可以在常温常压下按1,2-脱氢方式发生是一致的.
参考文献4
[1] Roithová J,Schrōder D.Selective activation of alkanes by gas-phase metal ions [J].Chem Rev,2010,110: 1170-1211.
[2] Schrōder D.Activation of methane by gaseous metal ions [J].Angew Chem Int Ed,2010,49: 850-851.
[3] Zhang D J,Liu C B,Bi S W,et al.A comprehensive theoretical study on the reactions of Sc+with CnH2n+2(n=1~3): structure,mechanism,and potential-energy surface[J].Chem Eur J,2003,9: 484-501.
[4] Sievers M R,Chen Y M,Haynes C L,et al. Activation of CH4,C2H6,and C3H8by gas-phase Nb+and the thermochemistry of Nb-ligand complexes [J].Int J Mass Spectrom,2000,195: 149-170.
[5] Armentrout P B.Activation of C2H6and C3H8by gas-phase Mo+: Thermochemistry of Mo-ligand complexes [J].Organometallics,2007,26: 5473-5485.
[6] Chen Y M,Armentrout P B.Activation of C2H6,C3H8,and c-C3H6by gas-phase Rh+and the thermochemistry of Rh-ligand complexes [J].J Am Chem Soc,1995,117: 9291-9304.
[8] Hanmura T,Ichihashi M,Kondow T.Dehydrogenation of simple hydrocarbons on platinum cluster ions[J].J Phys Chem A,2002,106:11465-11469.
[10] Heinemann C,Wesendrup R,Schwarz H.Pt+-mediated activation of methane: theory and experiment [J].Chem Phys Lett,1995,239: 75-83.
[11] 王丙星,朱井义,胡晓萍,等.过渡金属离子Pt+和甲烷气相反应机理的理论研究[J].原子与分子物理学报,2008,25(4): 745-749.
[12] Ye P,Ye Q,Zhang G B,et al.Potential energy surfaces and mechanisms for activation of ethane by gas-phase Pt+: A density functional study[J].Chem Phys Lett,2011,501: 554-561.
[13] Stephens P J,Devlin F J,Chabalowski C F,et al.Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields[J].J Phys Chem,1994,98: 11623-11627.
[14] Becke A D.Density-functional thermochemistry.III.The role of exact exchange[J].J Chem Phys,1993,98: 5648-5652.
[15] Lee C,Yang W,Parr R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1998,37: 785-789.
[16] Hay P J,Wadt W R.Ab initio effective core potentials for molecular calculations:potentials for the transition metal atoms scandium to mercury[J].J Chem Phys,1985,82: 270-283.
[17] Hay P J,Wadt W R.Ab initio effective core potentials for molecular calculations:potentials for potassium to gold including the outermost core orbitals[J].J Chem Phys,1985,82: 299-310.
[18] Couty M,Hall M B.Basis sets for transition metals: optimized outerpfunctions[J].J Comput Chem,1996,17: 1359-1370.
[19] Ehlers A W,Böhme M,Dapprich S,et al.A set of f-polarization functions for pseudopotential basis sets of the transition metals Sc-Cu,Y-Ag and La-Au[J].Chem Phys Lett,1993,208:111-114.
[20] Krishnan R,Binkley J S,Seeger R,et al.Self-consistent molecular orbital methods(XX):a basis set for correlated wave functions [J].J Chem Phys,1980,72: 650-654.
[21] Fukui K.Formulation of the reaction coordinate[J].J Phys Chem,1970,74: 4161-4163.
[22] Fukui K.The path of chemical reactions-the IRC approach [J].Acc Chem Res,1981,14: 363-368.
[23] Gonzalez C,Schlegel H B.Reaction path following in mass-weighted internal coordinates [J].J Phys Chem,1990,94: 5523-5527.
[24] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 09,Revision C.01[CP/DK].Gaussian,Inc,Wallingford CT,2010.
[25] Weast R C,Lide D R,Astle M J,et al.CRC handbook of chemistry and physics[M].89th ed.New York:CRC Press Inc,2009:10-204.
(责任编辑胡小洋)
Theoretical analysis for the structures and energetics on the reactionpathway of 1,2-dehydrogenation of propane by gas-phase Pt+
ZHAO Gaiyun,GUO Yue,ZHANG Ganbing,ZHONG Xinxin,HU Wei,XIA Qinghua
(Hubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Ministry of Education Key Laboratory
for the Synthesis and Application of Organic Functional Molecules, College of Chemistry and Chemical Engineering,
Hubei University,Wuhan 430062,China)
Abstract:The geometries,vibrational frequencies,and energetics of all stationary points including reactants,intermediates,transition states,and products on the 1,2-dehydrogenation pathway of the activation of propane by Pt+in gas phase were calculated by DFT method with UB3LYP functional combining with relativistic effective core potential for understanding its mechanism.Calculated results show that the whole reaction on 1,2-dehydrogenation pathway is exothermic by 195.7 kJ/mol.The rate-determining step is the step of the activation of the second C—H bond,which has a lower barrier of 48.7 kJ/mol.These mean that the 1,2-dehydrogenation of propane by gas-phase Pt+can take place readily at room temperature.These results agree well with the experimental observation.
Key words:activation of Propane;1,2-dehydrogenation pathway;gas-phase platinum ion catalysis;the reaction mechanism;density functional theory
中图分类号:O643.38
文献标志码:ADOI:10.3969/j.issn.1000-2375.2016.01.013
文章编号:1000-2375(2016)01-0069-04
通信作者
作者简介:赵改云(1989-),女,硕士生;张干兵,,副教授,E-mail: gbzhang@hubu.edu.cn
收稿日期:2015-05-29
