高效液相色谱-串联质谱法测定饲料中地克珠利的含量
2016-02-07侯轩朱聪英应永飞陆春波周炜张航俊
侯轩,朱聪英,应永飞,陆春波,周炜,张航俊
(浙江省兽药饲料监察所,杭州 310000)
高效液相色谱-串联质谱法测定饲料中地克珠利的含量
侯轩,朱聪英,应永飞,陆春波,周炜,张航俊
(浙江省兽药饲料监察所,杭州 310000)
建立了一种测定饲料中地克珠利的高效液相色谱-串联质谱方法。以乙腈提取样品中的待测物,正己烷除脂,离心后用高效液相色谱-串联质谱仪,选择多反应监测(MRM)、负离子模式进行定性和定量分析。结果表明,地克珠利在0.5~100 ng/mL浓度范围内线性关系良好,线性相关系数≥0.999,检出限为0.1 μg/kg,定量限为0.5 μg/kg。地克珠利在0.5、1和5 μg/kg三个添加水平下,三种饲料的平均加标回收率在79.2%~94.5%之间,相对偏差在1.7%~14.3%之间。本方法抗干扰能力强,操作简便,灵敏度高,适用于饲料中地克珠利的测定。
饲料;地克珠利;高效液相色谱-串联质谱
地克珠利(DIC)为类白色或淡黄色粉末,属于三嗪类抗球虫药,具有高效抗球虫能力,广泛应用于畜牧业中,尤其在防治鸡球虫病方面有着重大意义[1]。地克珠利具有低毒、广谱、用量小、无交叉耐药性等特点。2015年1月14日,欧盟发布法规(EU)2015/46,批准地克珠利作为鸡和兔饲料添加剂使用,预混合剂添加量为5 g/kg,且不应与其他抗球虫药混合使用。我国农业部公告第168号饲料药物添加剂使用规范中规定,地克珠利预混合剂每1 kg饲料中含地克珠利2 g或5 g,全饲料添加量为1 g/1000 kg,蛋鸡产蛋期禁用。在国内,地克珠利作为兽用药物从1996年开始大规模使用[2],但是在地克珠利应用的过程中,滥用药物、不遵守休药期等不合理的使用情况较为严重,易引发球虫耐药性,带来一系列兽药残留问题。因此从源头出发,对饲料中地克珠利的含量进行监测显得势在必行。
目前对地克珠利检测研究多有报道。针对鸡肉样品,施祖灏等[3]建立了鸡组织中地克珠利和妥曲珠利残留HPLC检测方法,苏晓红等[4]利用MISPE技术,采用高效毛线管电泳技术建立鸡肉中地克珠利的残留分析方法;De等[5]建立液相色谱法对饲料中地克珠利进行检测,Mortier L等[6]、赵岳等[7]均选择固相萃取装置对饲料样本进行净化,液质联用法进行检测。本研究针对三种饲料样本,对仪器条件及前处理方法进行优化,成功建立了一种快速有效监测饲料中地克珠利含量的高效液相色谱串联质谱检测方法。
1 材料与方法
1.1 实验仪器 高效液相色谱仪(LC 30A,岛津公司)、串联三重四极杆质谱仪(TRIPLE QUAD 4500, ABSCIEX公司);电子天平(XS-205,Mettler公司);电子天平(SL502 N,上海民桥精密科学仪器有限公司);氮吹仪(N-EVAP,Organomation公司);冷冻离心机(3-18k ,Sigma公司);振荡器(ZD-8800,华立达公司)。
1.2 药品与试剂 地克珠利标准品( H0130805,含量≥99.7%,100 mg,购于中国兽医药品监察所),甲醇、甲酸、乙腈、N,N-二甲基甲酰胺(色谱纯,购于默克公司),乙腈(分析纯,购于上海凌峰化学试剂有限公司)。
1.3 溶液配制
1.3.1 地克珠利标准贮备液的配制 准确称取适量地克珠利标准品,用N,N-二甲基甲酰胺溶解配制成100 μg /mL,4 ℃保存,有效期1个月。
1.3.2 地克珠利标准工作溶液的配制 准确量取适量标准储备溶液,用甲醇稀释成10 μg/mL的标准工作溶液,4 ℃保存,有效期1个月。1.3.3 地克珠利基质匹配标准曲线工作液的配制 分别取空白配合饲料、预混合饲料和浓缩饲料按样品前处理方法进行处理后,在洗脱溶液中加入适当体积标准工作溶液混合,使之成0.5、1.0、10、50、100 ng/mL基质匹配标准上机液,现配现用。
1.4 方法
1.4.1 仪器条件
1.4.1.1 色谱条件 用十八烷基键合硅胶色谱柱(Waters C18 3 μm, 3.0×150 mm),以0.2%甲酸水溶液为流动相A,乙腈为流动相B,按表1进行梯度洗脱,流速为0.3 mL/min,柱温40 ℃,进样量5 μL。

表1 梯度洗脱程序表
1.4.1.2 质谱条件 电喷雾离子源;负离子扫描,多反应监测模式(MRM),离子源电压:4500 V;碰撞气能量:高;辅助加热气温度:500 ℃;气帘气流速:10 L/h;离子源气流流速:Gas1 为50 L/h,Gas2为50 L/h。目标化合物MRM参数见表2。
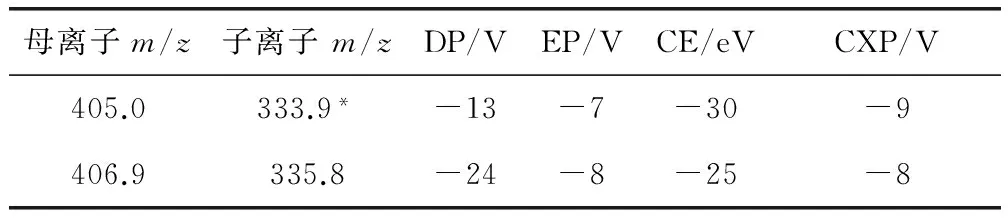
表2 地克珠利多反应监测条件
DP:去簇电压,EP:碰撞室入口电压,CE:碰撞能量,CXP:碰撞室出口电压,333.9*为定量离子
1.4.2 样品前处理 称取样品2 g(精确到0.05 g)于50 mL离心管中,加入15 mL乙腈,300 r/min振荡提取10 min,10000 r/min离心10 min,转移上清液至另一50 mL离心管中,残渣用15 mL乙腈重复提取一次,合并两次上清液,50 ℃氮气吹干。加入0.2%甲酸乙腈和正己烷各2 mL,涡旋1 min,10000 r/min离心取下层,过0.22 μm微孔滤膜后液相色谱-串联质谱测定。
1.4.3 基质匹配标准曲线绘制 取空白配合饲料、预混合饲料和浓缩饲料按1.3.3项处理,所得溶液过0.22 μm有机滤膜,HPLC-MS/MS分析。以药物浓度为横坐标,地克珠利定量离子峰面积为纵坐标,绘制标准曲线。
1.4.4 样品添加回收率、准确度和精密度测定 分别对配合、预混合和浓缩饲料进行0.5、1.0、5.0 μg/kg的药物空白添加回收实验,按1.4.2项前处理后进行HPLC-MS/MS检测。每个浓度设5个平行,连续做3 d,计算添加回收率、日内变异系数和日间变异系数。
2 结果与分析
2.1 质谱条件优化 为了获得更高的离子强度,在MRM模式下优化质谱条件,确定了地克珠利在多反应监测模式下信号采集的特征离子对,具体监测条件列于表2。
2.2 液相色谱条件优化 为了获得较好的色谱峰型,本文对色谱柱及流动相进行了优化。采用1.4.1.1项所述色谱柱及流动相条件,色谱行为良好,能满足检测要求。从标准品、加标样品的MRM色谱图(图1~图3)可见,在待测药物出峰的位置没有杂质干扰。
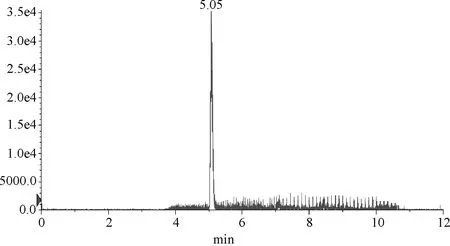
图1 标准品的MRM图(浓度1.0 μg/L)
2.3 标准曲线、检出限及定量限 按1.4.2项处理三种空白饲料样品后,高效液相色谱串联质谱检测,以峰面积作为纵坐标,浓度为横坐标绘制标准曲线。由表3可知,在浓度为0.5~100 ng/mL浓度范围内,线性关系良好,r>0.999,取信噪比S/N=3的样品浓度为方法检出限(LOD),取信噪比S/N=10 的样品浓度为方法定量限(LOQ)。
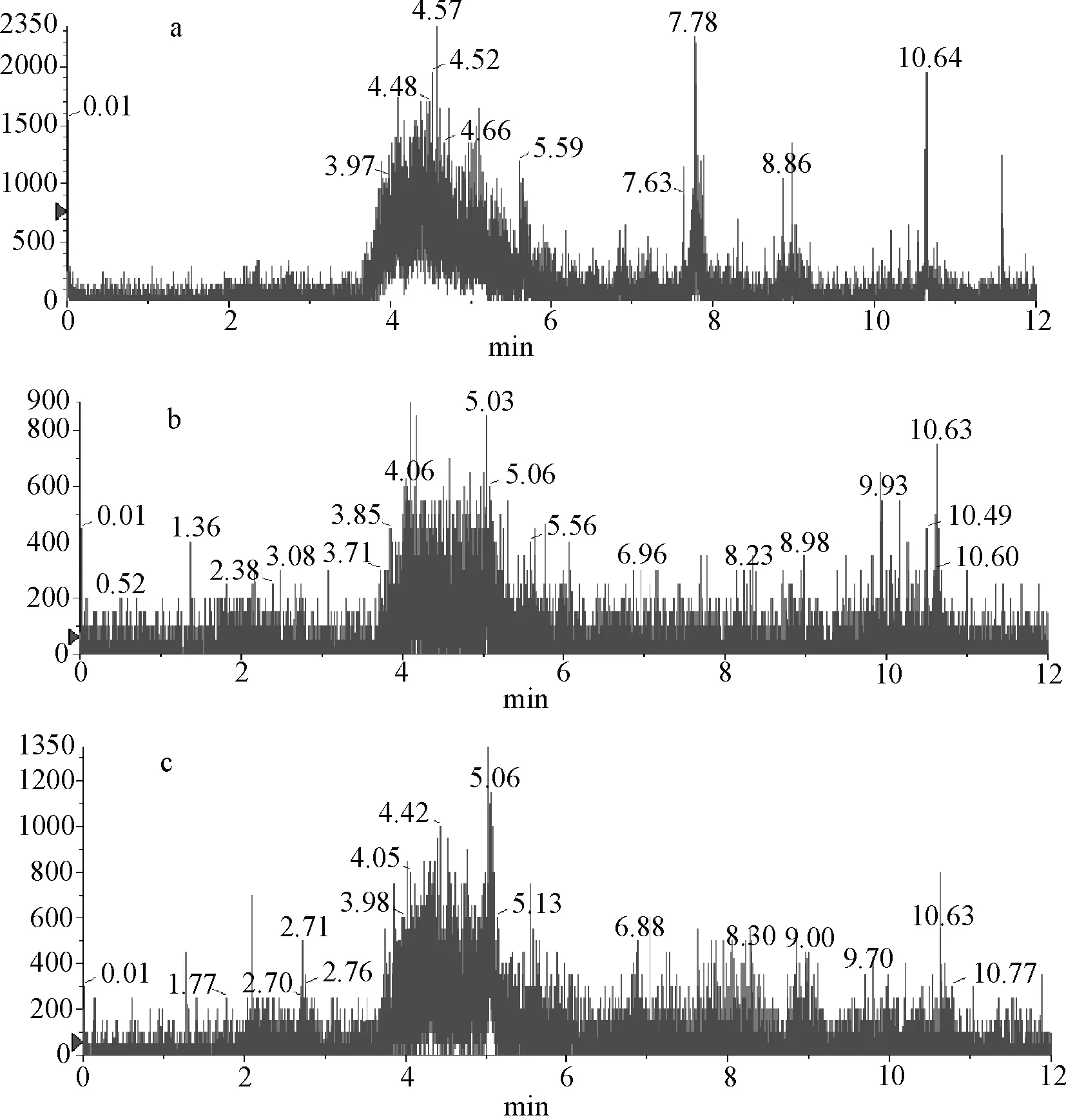
a:配合饲料 b:预混合饲料 c:浓缩饲料图2 空白饲料的MRM图
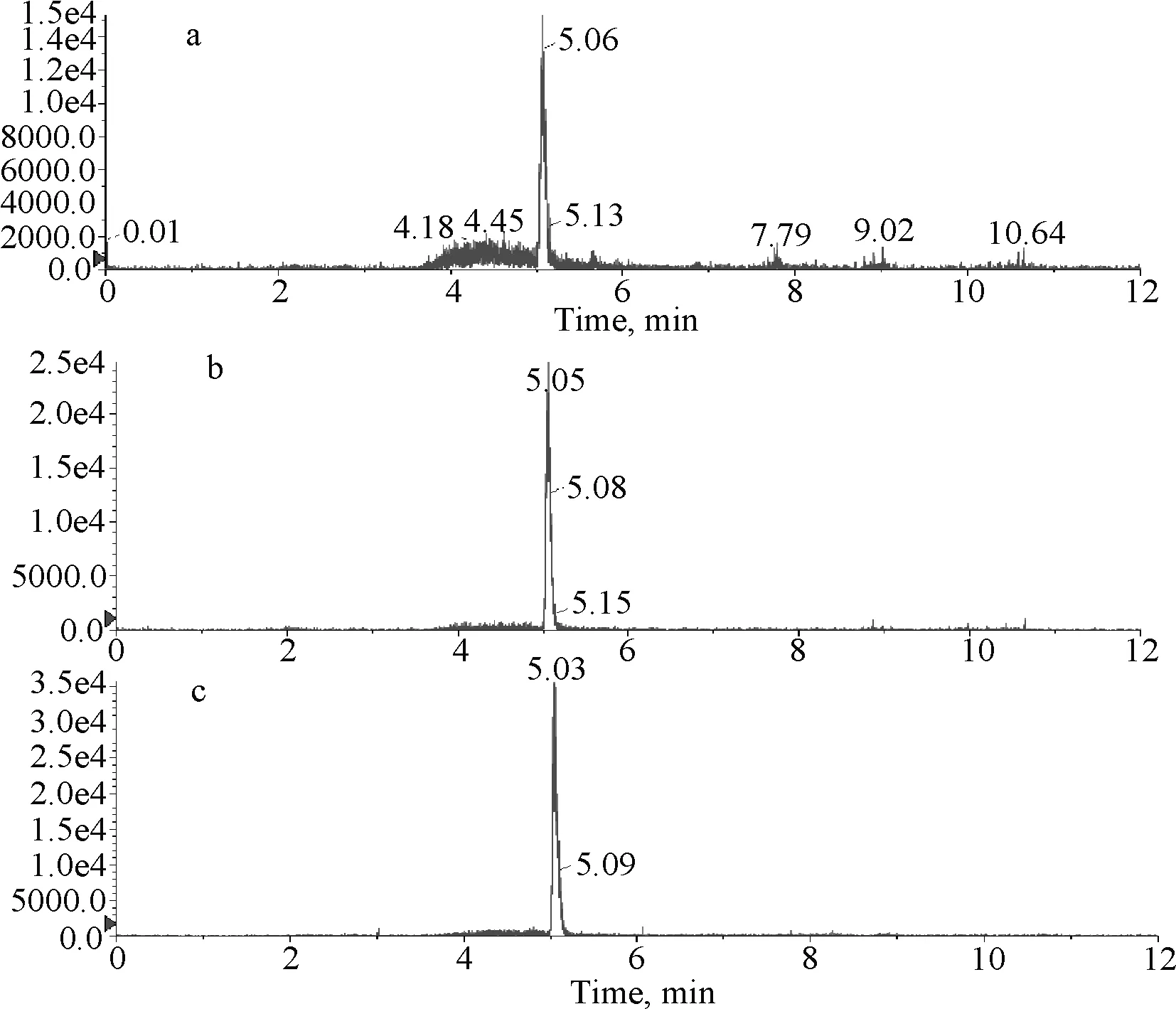
a:配合饲料 b:预混合饲料 c:浓缩饲料图3 空白饲料添加样品的MRM图(地克珠利浓度1.0 μg/kg)
2.4 方法的回收率、精密度 在0.5、1.0、5.0 μg/kg浓度添加水平下对配合、预混合和浓缩饲料进行添加回收实验,每个浓度设5个平行,连续做3 d,计算添加回收率、日内变异系数和日间变异系数,结果见表4。
由表4可见,配合饲料、预混合饲料、浓缩饲料中,地克珠利平均回收率在79.2%~94.5%之间,日内变异系数、日间变异系数均小于15%,方法回收率高,精密度好。
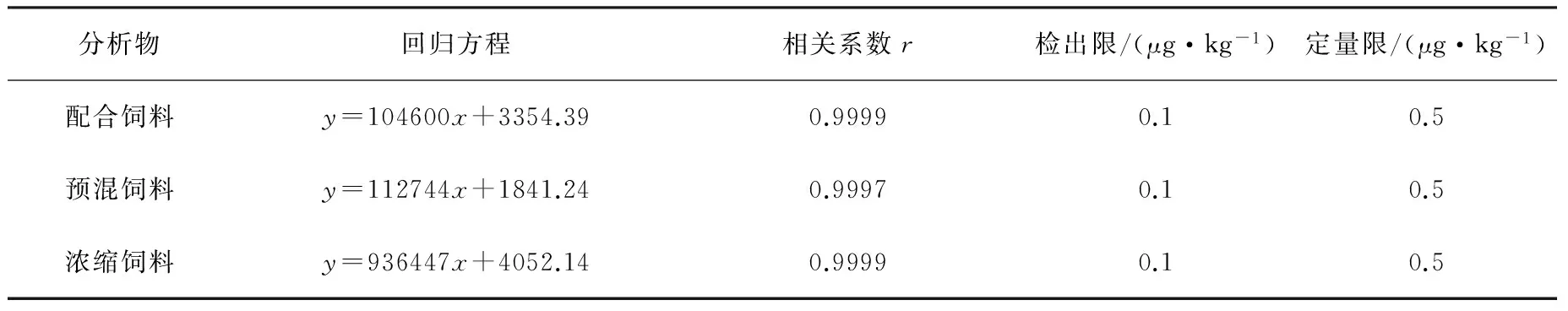
表3 配合饲料、预混合饲料和浓缩饲料中地克珠利基质匹配标准曲线及检出限和定量限
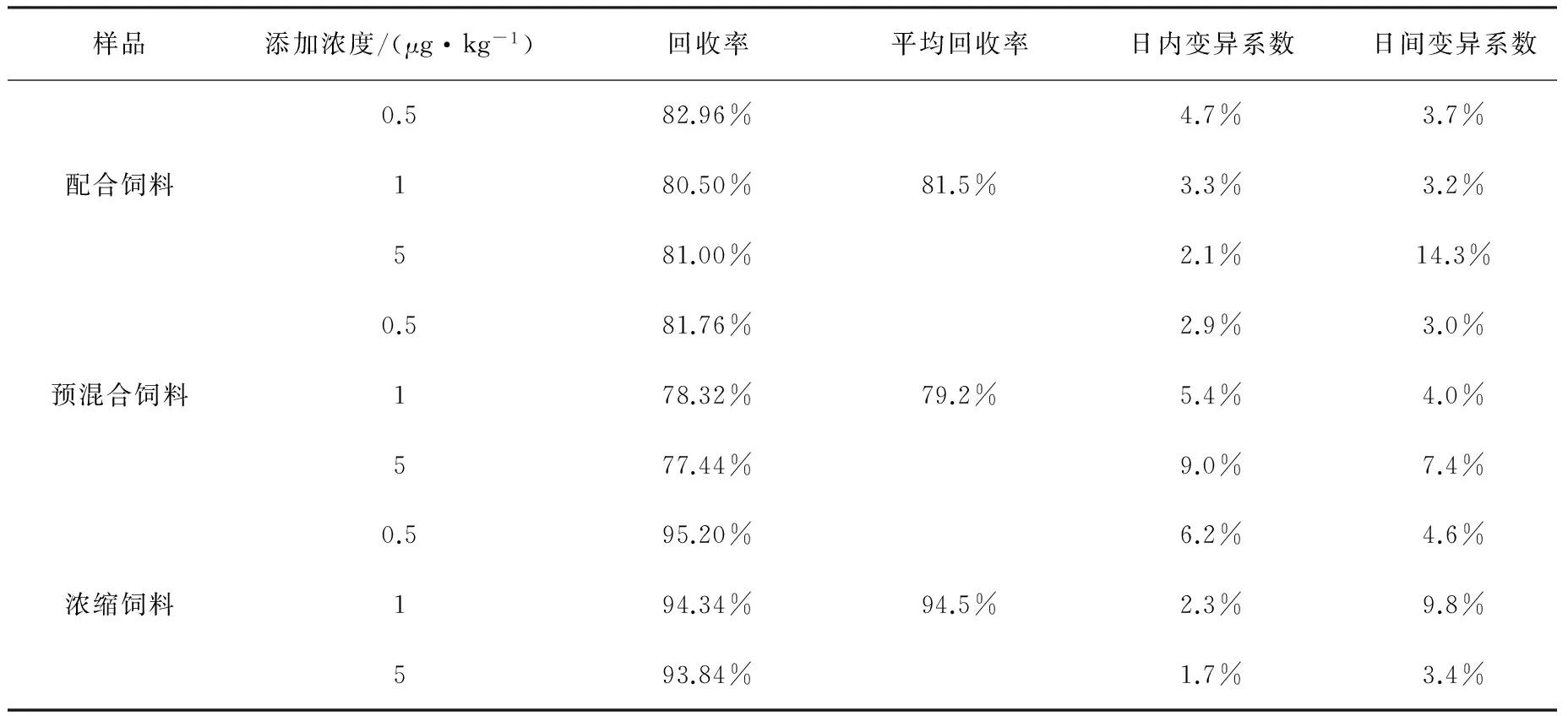
表4 地克珠利回收率及日内、日间变异系数
3 讨论与小结
3.1 色谱条件优化 地克珠利色谱条件在多篇文献中均有研究。杨丽萍等[8]选用乙睛∶0.1%乙酸(65∶35,V/V)等度洗脱,Ai等[9]选用乙睛∶0.1%乙酸(55∶45,V/V),流速为等度洗脱,罗浩师等[10]选用乙睛∶0.1%甲酸梯度洗脱,刘永涛等[11]选用乙腈∶0.3%乙酸(45∶55,V/V)等度洗脱,考虑到地克珠利离子化效率以及快速检测目的,本试验对色谱流动相进行了研究,经过优化条件,最后采用乙腈-0.2%甲酸水按表1梯度洗脱,地克珠利在5 min左右出峰并且峰形尖锐,达到良好的洗脱效果。
3.2 定量、定性离子的确定 根据欧盟对残留鉴定的技术文件规定,采用液质联用法确证动物源食品中的残留物质需要获得4个鉴定分(1个母离子为1.0分,1个子离子为1.5分)。地克珠利的分子结构中含有3个同位素氯,具有多个明显的氯离子同位素峰,根据各天然同位素所占比例的不同,试验最终选择丰度最高的m/z405.0和m/z406.9两个离子为母离子,之后通过对碰撞能量和碰撞室出口电压的选择优化,选择m/z333.9和m/z335.8两个子离子分别作为各自的子离子,由于m/z333.9响应值较高,因此作为地克珠利的定量离子。这样共获得5个确证分,满足欧盟对残留物的确证检测要求。
3.3 样品前处理方法的优化 Mortier L等[6]采用0.1%甲酸乙腈提取鸡肉和饲料基质中的样品,浓缩提取后,用内标法进行定量;赵岳等[7]在测定鸡饲料中地克珠利含量时,采用硅胶固相萃取柱进行净化后上机检测。本实验经过研究,省去固相萃取净化步骤,而是在样品复溶的同时加入正己烷进行提脂净化,最后取下层澄清溶液上机检测,结果可见空白样品基线清晰,加标样品峰形锐利且无其他杂峰干扰,采用外标法定量,回收率在79.2%~94.5%之间,能够满足检测要求。
本实验建立的饲料中地克珠利HPLC-MS/MS检测方法,地克珠利的回收范围为79.2%~94.5%,日内变异系数范围为1.7%~9.0%,日间变异系数范围为3.0%~14.3%,检测限为0.1 μg/kg,定量限为0.5 μg/kg。该方法优化了前处理条件,操作简便,检测方法灵敏度高,重现性强,符合兽药残留分析性能要求,可用于日常样品分析。
[1] 周变华, 王宏伟, 薛飞群, 等. 地克珠利抗鸡球虫作用机理研究进展[J]. 中国家禽, 2011, 33(21): 40-42.
[2] 才凤峰. 鸡球虫病的防治—抗球虫药的使用[J].中国家禽, 2004, 26(6): 30-34.
[3] 施祖灏, 朱良强, 卢运站, 等. 鸡组织中地克珠利和妥曲珠利残留 HPLC 检测方法的建立[J]. 中国兽医学报, 2009, 29(1): 79-81.
[4] 苏红晓. 动物源性食品及环境中抗球虫药物残留分析技术的研究[D]. 合肥:安徽农业大学, 2013.
[5] Jan De Kock, Maurits De Smet, Rudy Sneyers. Determination of diclazuril in animal feed by liquid chromatography[J]. Journal of chromatography A, 1992, 606(1): 141-146.
[6] Leen Mortier, Els Daeseleire, Carlos Van Peteghem. Determination of the coccidiostat diclazuril in poultry feed and meat by liquid chromatography-tandem mass spectrometry[J]. Analytica chimica acta, 2005, 529(1): 229-234.
[7] 赵岳,沈建忠,程林丽,等. 超高效液相色谱-串联质谱法测定鸡饲料中地克珠利的含量[J]. 中国饲料, 2012,(3):36-39.
[8] 杨丽萍,高淑霞,孙海涛,等. 液相色谱-串联质谱法研究地克珠利在家兔组织中残留及消除规律[J]. 畜牧与饲料科学, 2011, (12): 6-9.
[9] Lianfeng Ai, Hanwen Sun, Fengchi Wang,etal. Determination of diclazuril, toltrazuril and its two metabolites in poultry tissues and eggs by gel permeation chromatography-liquid chromatography-tandem mass spectrometry[J]. Journal of chromatography B, 2011, 879(20): 1757-1763.
[10]罗浩师,张丽芳,薛飞群. 高效液相色谱-串联质谱法测定鸡脂肪中地克珠利残留量[J]. 中国家禽, 2012, 34(20): 14-17.
[11]刘永涛,艾晓辉,李乐,等. 超高效液相色谱法测定鱼体组织中地克珠利残留量[J]. 分析试验室, 2014,(4):420-423.
(编辑:侯向辉)
Determination of Diclazurilin Feeds by High Performance Liquid Chromatography-Tandem Mass Spectrometry
HOU Xuan,ZHU Cong-ying,YING Yong-fei,LU Chun-bo,ZHOU Wei,ZHANG Hang-jun
(ZhejiangProvincialSupervisoryInstituteofVeterinaryDrug,Hangzhou310000,China)
An analytical HPLC-MS/MS method was established for determination of diclazuril in feeds. The samples were extracted with acetonitrile, then defatted with n-hexane. The treated samples were analyzed by HPLC-MS/MS after centrifuged. The analysis was performed with Multi Reaction Monitor(MRM) in negative ion mode. The results indicated good lineartites in the concentration range from 0.5 to 100 ng/mL for diclazuril, the correlation coefficient was over 0.999. The limit of detection(LOD) and the limit of quantitation(LOQ) were 0.1 and 0.5 μg/kg,respectively. While the spiked contents of diclazuril standard at 0.5,1,5 μg/kg, the recoveries and relative standard deviations(RSD) were in range of 79.2%~94.5% and 1.7%~14.3%,respectively. With the advantages of good anti-interference ability, rapidness and sensitivity, the method adapted to the determination of diclazuril in feeds.
feeds; diclazuril; HPLC-MS/MS
侯轩,硕士研究生,从事兽药和畜产品质量安全相关检测研究。E-mail:zjmy607@sina.com
2016-01-08
A
1002-1280 (2016) 04-0057-05
S859.2
