老年人身体功能退行性变的机理研究新进展
2015-12-29陈福军
陈福军 杨 泽
2.北京医院 北京老年医学研究所 卫生部老年医学重点试验室 100730
国家科技部十二五支撑计划(2012BAI10B01)
老年人身体功能退行性变的机理研究新进展
陈福军1杨泽2※
2.北京医院北京老年医学研究所卫生部老年医学重点试验室100730
国家科技部十二五支撑计划(2012BAI10B01)
【摘要】最近的研究发现了端粒介导衰老的一些新机制“不同的通路…交叉和汇合到线粒体的模型”,即:端粒缩短和相关的DNA损伤反应,会促使线粒体功能障碍的发生,减少氧化损伤的防御,并破坏ATP的产能过程。这可以解释干细胞、祖细胞和有丝分裂后组织的能量生成,普遍下降的现象。
【关键词】端粒DNA损伤反应线粒体功能障碍衰老
端粒维护一直被证明与衰老有关。体外培养的人类成纤维细胞经有限次数的分裂,进入到不分裂状态,即衰老(senescence)。后来证实在染色体的末端存在有一特殊的DNA序列片段,这一片段通称为端粒,端粒在每一次复制之后都会变得越来越短。根据这些观察结果,人们猜测,端粒的逐渐丢失、变短,可能代表了某一种驱动衰老的分子钟。针对端粒序列的研究强调了DNA完整性的重要意义,因为研究者已经确认端粒的异常等同于DNA损伤, 由于DNA损伤将会激活DNA损伤反应通路,并导致p53基因的激活。 然后,p53将会诱导干细胞和祖细胞的生长停滞,产生细胞衰老和凋亡(apoptosis)。
1.端粒与衰老
端粒完整性,作为一条单独涉及长寿的主要调节因子的线索,人们已经进行了广泛的研究。端粒是TTAGGG重复序列,作为染色体帽可防止染色体末端被识别为DNA,产生损伤。大多数人类细胞缺乏适当水平的端粒酶来维护端粒,这导致随着每一次复制后端粒出现缩短。Hayflick和Moorhead最初在1960年进行了人类纤维原细胞的开创性研究,他们观察到这些细胞的体外分裂次数有限,经连续传代端粒缩短,并最终导致衰老,由此推断端粒长度在衰老中有重要作用。相反,激活端粒酶可使端粒延长,并这使成纤维细胞可以绕过衰老无限增殖,证明了端粒缩短和细胞衰老之间有因果关系[1]。事实上,在人体许多组织中包括增殖部分和静态组织,端粒长度均表现为随年龄增长而逐渐变短的特征。
有趣的是,即使细胞有端粒酶表达,但随着时间的推移端粒依然缩短,这表明端粒长度的调控存在有复杂的机制。此外,许多研究已经发现人类外周血白细胞的端粒缩短和典型老龄相关疾病的发病风险之间存在正相关性。来自维持端粒长度至关重要基因的丧失功能(loss of function, LOF)突变患者,提供了端粒对衰老影响的进一步支持性证据,因为这些突变使这些个体更易加速衰老(早老)。研究者在先天性早老综合症角化不良患者中,发现了TERC(端粒酶的RNA组分)和TERT(端粒酶的催化组分)基因的突变[2]。在编码沃纳(Werner)综合征ATP依赖解旋酶(WRN)和共济失调毛细血管扩张症中(ATM)突变的基因,分别导致了沃纳综合征和神经退行性疾病共济失调毛细血管扩张症[3]。除了这些多系统疾病,功能丧失的TERC和TERT 基因的LOF突变与多个器官受限疾病的发生有关,如肝纤维化、特发性肺纤维化和骨髓衰竭综合征。端粒相关退行性表型出现与否取决于端粒功能障碍的程度,在先天性角化不良患者的后代中,端粒越短者其病状出现的越早,其预后也越严重[4]。虽然端粒维护障碍的疾病研究,为我们提供了端粒对于器官完整性和寿命调控重要性的证据,但是鉴于在正常衰老中,通常见不到这些患者才有的病理改变,因此对待这些研究结果要谨慎,不能简单外推用来解释正常的衰老过程。这些患者的衰老恶化表型可能与多个增殖和静态组织中端粒过度缩短有关,其过度缩短超出在人类正常衰老过程中所看到的端粒缩短,当然不排除端粒病态缩短也可能受环境因素驱动。
端粒与衰老之间的联系也已经从小鼠研究中得到证明。人们逐渐认识到,野生型小鼠衰老过程中的端粒长度和完整性均受到损害[5]。有趣的是,只要在细胞中存在有一个或几个功能失调的端粒,似乎就足以引发DNA损伤反应,而这可能会导致发生某些疾病的病理改变[6]。因此,可以理解端粒酶的过表达,可延缓抗肿瘤的基因工程鼠的某些年龄相关的退行性变化[7]。此外,在缺乏端粒酶活性的小鼠中证实了端粒对机体健康和寿命的影响,其产生了多个与年龄相关的退行性表型,一旦他们的端粒变短,死亡就会更早发生。此外,只要端粒变短,沃纳综合征和共济失调毛细血管扩张症的小鼠模型就会发生典型的人体病变,这证明短端粒在疾病表型发生中的重要作用[8]。最后,端粒酶过表达可以逆转小鼠多个组织中与年龄相关退行性变的表型[9]。
这些研究表明,端粒功能障碍可以促使组织功能下降,加速衰老,使寿命缩短。更重要的是提示我们,衰老过程是可以预防的,甚至可被重新激活的端粒酶逆转。然而,很明显,关于自然衰老中端粒的确切作用,以及端粒如何影响与年龄相关的病理变化,目前还只有很少和基本的了解。
2.端粒-线粒体的连接
端粒变短如何引起广泛的退行性变?一个线索来自于对先天性角化不良、沃纳综合征和共济失调毛细血管扩张症患者以及对小鼠的观察,当端粒功能失调时,发生了器官功能衰竭,特别是在高增殖性器官中,如肠道、皮肤和骨髓。因为这些器官依赖驻留其内部的干细胞和祖细胞介导的连续再生的功能。这一观察结果已导致“端粒基础上衰老”的假说,主要内容是端粒缩短激活p53,引起干细胞缺陷和诱导干细胞生长停滞,衰老和凋亡[10]。确实,在这些细胞中,伴随着端粒缩短,引起了p53活性增加,并导致了高水平的凋亡。
然而,在缺乏p53或其下游靶蛋白的小鼠中观察到,造血系统、皮肤和胃肠道及病变组织伴随有器官组织内的干细胞和祖细胞发挥着自救功能。虽然衰老的干细胞功能丧失的理论有助于理解需要高度再生组织产生的衰竭,但是它不容易解释在为维持组织稳态的静态组织中,干细胞和祖细胞的活性较低,以及其在静态组织中的年龄依赖性变化。
事实上,大多数有丝分裂后的组织,如心脏、肝脏和胰腺发生的全身代谢紊乱、机能衰退和衰老,人们公认的特征是,患者的端粒维护障碍和端粒的功能失调[11]。例如,对于先天性角化不良、沃纳综合征和共济失调毛细血管扩张症的个体是极易发生胰岛素抵抗和糖尿病。此外,经端粒功能失调小鼠和共济失调的小鼠模型验证,先天性角化不良会引起患者的心肌病。肝纤维化和肺纤维化代表先天性角化不良患者的病理生理表现。再生障碍性贫血进行骨髓移植的细胞毒性化疗时,产生的肝毒性是先天性角化不良患者的主要副作用。
静态组织如心脏和肝脏也已经报道有年龄依赖性端粒缩短,然其机制不清。总之,这些结果提示端粒引起的衰老有额外的附加机制。超出了传统p53依赖检查点的细胞凋亡和衰老反应。最近对有端粒功能障碍的TERT缺陷小鼠的研究工作,发现其他机制的线索已经浮出水面。研究表明在多种组织中出现线粒体生物合成和功能的显著性损伤,包括肝脏、心脏和造血干细胞,这可能提出了能量维护中的一个基本问题,结果可能有助于解释这些小鼠的早老表型。这些标记线粒体的变化,似乎是由转录辅激活辅因子PGC1α和PGC1β及其下游靶分子(图1)的联合抑制引起的。这是通过p53直接结合到PGC1α和PGC1β基因启动子上介导的;因此,缺乏p53基因的端粒障碍小鼠,PGC的表达正常,线粒体DNA(mtDNA)含量增加,糖异生增加并减少了阿霉素引起的心肌病。
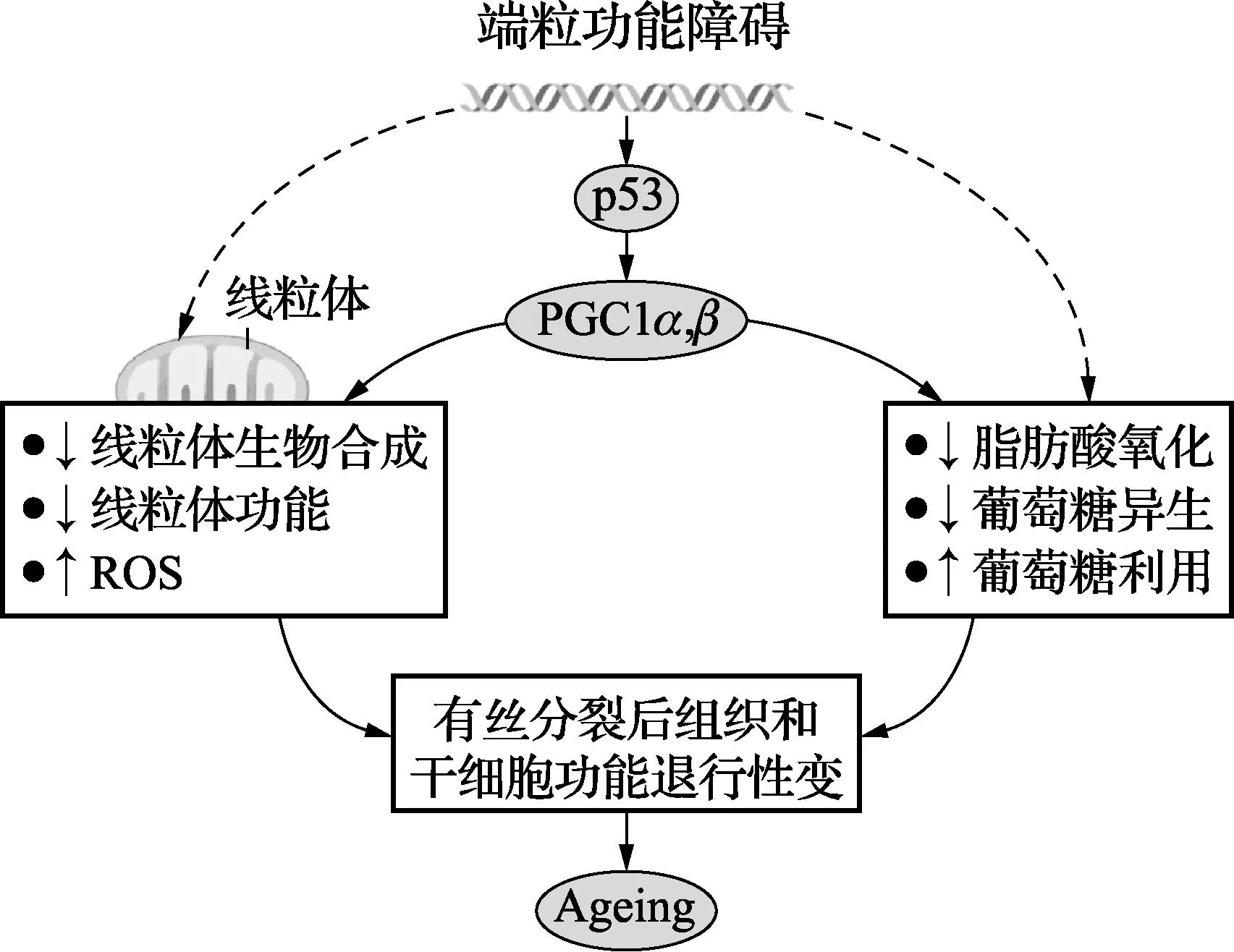
图1 端粒-p53-PGC通路
端粒功能紊乱促使p53与PGC1α和PGC1β的启动子相结合并抑制了PGC1A和PGC1B的表达。两个辅激活因子的抑制,损伤了线粒体整体生物合成和功能,并导致有缺陷的ATP生成和ROS水平增加。PGC也通过调节不同的生化通路如脂肪酸氧化、糖异生、葡萄糖摄取和氧化,参与能量代谢。线粒体功能等生化通路的退行性变可能同样导致组织干细胞和有丝分裂后的组织功能下降,驱动了衰老。端粒酶激活或PGC过表达均可逆转PGC相关的代谢和端粒功能障碍小鼠的线粒体变化。端粒功能障碍还可导致通过其他通路(虚线箭头)引致线粒体功能和能量代谢的损害。
在调节多种过程中PGC线路有重要的影响,TERT缺陷小鼠显示有糖异生,β氧化和ROS防御必需的基因表达降低,并且引致OXPHOS受到很大的损害, ATP生成降低,糖异生的能力受损,与年龄相关的心肌病发生。值得注意的是,这些改变更加显著地增加端粒功能障碍。重要的是,端粒功能紊乱小鼠的TERT或PGC1α过表达改善了线粒体呼吸和糖异生,这证明了抑制PGC 产生了TERT缺陷小鼠的表型。这一端粒和线粒体的链接也得到其他研究的证明,表明包括培养的人成纤维细胞,TERT基因突变体过表达的成纤维细胞,TERT缺陷小鼠心脏组织内的ROS水平增加和线粒体功能障碍。这些缺陷的基础,是继发于p21转化生长因子β(TGFβ)与p53通路的基因激活和线粒体损伤增加[12]。此外,一项新近的端粒功能失调小鼠研究发现,线粒体膜超极化减少,和Ca2+内流受损,会导致β-细胞释放胰岛素的下降[13]。
先前的研究已经表明,TERT具有端粒延长的独立作用。然而,TERC基因缺陷小鼠缺乏端粒酶活性,但有完整的TERT表达,显示了PGC在线粒体抑制中的核心作用,这表明,端粒功能障碍是驱动这些变化的决定性因素[14]。此外,Tert-和Terc-基因敲除小鼠表型没有区别,并有相似的转录谱95。这就是说,研究这些基因的目的,不是要严格地排除Tert-的端粒的独立作用。事实上,持续研究是必要的,因为已知TERT定位于线粒体,保留有逆转录酶活性,执行着不同的线粒体功能(如调制线粒体DNA的完整性,改善呼吸链功能和影响ROS产生),并潜在地激活其他通路,如WNT通路[15]。此外,衰老组织常常表现伴有端粒功能障碍,p53活性和DNA损伤增加以及PGC水平和线粒体功能降低。
重要的是,来自患者和小鼠模型的细胞研究证实了端粒和线粒体之间的联系。例如,来自沃纳综合征患者细胞的线粒体功能受到损害且ROS水平增加[16]。虽然这些观察结果支持端粒-p53-线粒体衰老轴的重要性,但是更多的工作是必需的,以评估在人体端粒缩短的条件下,是否线粒体生物合成和功能会持续受损。
3.衰老的整合模型
联合端粒和线粒体二合一的衰老模型,支持由DNA损伤-诱导p53激活,经线粒体生物合成和功能的主调节因子,PGC1α和PGC1β的抑制,导致线粒体功能障碍的观点。端粒-p53-线粒体的衰老模型整合了很多衰老过程中的重要因子。在基因组水平上, DNA损伤驱动衰老。DNA损伤可能源于端粒缩短或源于介导DNA稳定性和DNA修复基因的表达减少。其次,该模型考虑了高反应TP53等位基因小鼠的衰老综合症, 小鼠模型显示DNA损伤增加,是由于TERC,Tert, DNA修复基因Ku80蛋白(也称为XRCC5), 乳腺癌1(BRCA1)和Zmpste基因的突变,编码金属蛋白酶参与核纤层蛋白A(lamin A,是核膜的一个重要组成部分),而金属蛋白酶突变引起早老[17]。最后,该模型考虑了衰老表型,源于线粒体功能障碍,因为缺乏PGC1α,PGC1β,BMI(p16基因的负调节子,在许多组织中被上调)或FOXO的小鼠加速发生组织退行性变和线粒体功能障碍[18]。
这种模式可能也解释了缓慢但渐进的生理性退行性变和衰老过程的本质(图2)。受前馈循环DNA损伤的影响, 随后发生的线粒体功能障碍,增加的ROS水平,和其他可能的线粒体来源的因子,如铁 - 硫(Fe-S)原子簇和NADH / NAD比值,进一步引起DNA损伤。这些促进ROS水平生成,加剧了基因毒性损害的循环,特别是损害了端粒的G 富集序列,继而持续激活p53基因,进一步促使线粒体功能下降,并产生了更多的ROS。ROS水平增加也会损害其他细胞成分,包括线粒体DNA,进一步维持这种前馈循环的损坏,抑制线粒体DNA编码氧化磷酸化(OXPHOS)基因的表达。在核DNA或线粒体DNA严重损伤的条件下,但是,可能有旁路循环替代,如报道的突变小鼠那样(携带Polγ的突变),其早老表型可源于不同组织中的细胞凋亡增加[19]。
4.衰老的机制和理论
在衰老模型中,由于端粒缩短,受损的DNA修复和ROS的水平增加,引起DNA的损伤增加,激活p53基因,高水平表达的p53通过抑制PGC1α和PGC1β(促进线粒体生物),最终导致线粒体功能损伤。
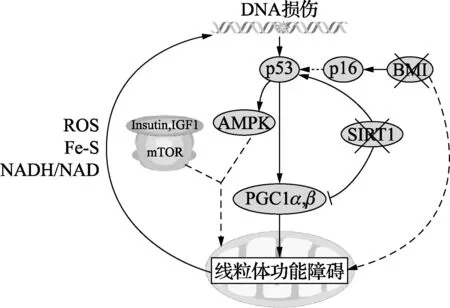
图2 衰老的整合理论
在衰老模型中,由于端粒缩短,受损的DNA修复和ROS的水平增加,引起DNA的损伤增加,激活p53基因,高水平表达的p53通过抑制PGC1α和PGC1β(促进线粒体生物),最终导致线粒体功能损伤。p53介导的线粒体功能障碍通过影响ROS, Fe-S簇和NADH / NAD的产生,启动了DNA损伤环路,接着又进一步导致p53激活和线粒体的退行性变。这种前馈回路也解释了在衰老过程中很多分子有不同,甚至有相反的作用。这里所示的几个组分(p53,线粒体和AMPK)在轻微应激条件下,证明有保护细胞的功能,但在更严重的应激条件下,则呈现促进细胞衰老的作用。p53和其他已经证明涉及衰老通路的分子之间有相互作用。 p53抑制哺乳动物胰岛素和IGF1通路和mTOR通路分子靶点的活性,并激活AMPK。但是这些通路的活性改变,如何改变线粒体功能并增加DNA损伤促进了老化过程是不明确的。其他p53依赖和p53独立的通路可能共同诱导着线粒体功能障碍。例如,BMI1间接抑制p53的激活,并且BMI1通过与MDM2(p53的负调节因子)的相互作用,丧失了上调p16蛋白的表达,间接增加了p53活性(虚箭头)。 BMI1也已显示可(间接地)诱导线粒体功能障碍。此外,沉默调节蛋白的缺失也可能导致线粒体功能障碍,如SIRT1的激活降低了p53的活性,SIRT1的抑制促进p53激活和其下游功能。 SIRT1也激活PGC1α,从而提高线粒体生物合成。线粒体功能障碍的后果是,当轻度功能障碍(如,ATP生成和β氧化降低)时,没有细胞损失。然而,在重度应激条件下,线粒体功能障碍导致器官功能障碍并伴随衰老和凋亡的增加,促使实体组织的丢失[19]。
这里提出的模型,仍有待确定参与介导线粒体和代谢及其他通路的存在,如有端粒失调的p53缺失小鼠只能够部分恢复PGC的水平和部分改善线粒体缺陷。其他p53家族成员是首要候选因子,特别是p63蛋白,已被证明参与生物体和细胞的衰老[20]。沉默调节蛋白(Sirtuins)可能具有一定的作用,因为它们与端粒相关联,并调节p53和PGC1α的表达[21]。事实上,已经发现在老化小鼠和人衰老组织中SIRT1的活性减少,而这可能有助于增加p53活性和抑制PGC1α活性。然而,最近的报告质疑SIRT1调节寿命的作用[22],并强调需要着重在Sirtuins和衰老轴的联动作用及其与年龄相关病变的关联上开展研究。另一种潜在的候选因子是BMI-p16基因通路,因为它与衰老高度关联,并且BMI的缺失引起了线粒体功能的损伤。
最后,已经提出p21依赖信号可诱导培养中端粒缩短到临界水平的人成纤维细胞的线粒体功能障碍,但应注意的是,成纤维细胞较少依赖线粒体和OXPHOS产生ATP,其主要经糖酵解生成ATP。在这种情况下,必须指出,端粒功能失调的酵母展示了OXPHOS基因表达增加和线粒体数量的增生,且有报道衰老的成纤维细胞端粒功能失调,有增加线粒体生物合成的作用[23]。这些研究突出的不仅是端粒功能紊乱对线粒体生物学有细胞水平的特异性作用,而且还有小鼠和酵母种属之间的差异,这可能与生长条件和端粒功能紊乱后的酵母特异性调节有关。
5.结论
在这方面,重要的是必须要明确其他线粒体生化通路受损(除外ROS产生和OXPHOS)也可能参与细胞和生物体衰老的机制。解读这些衰老通路的网络,可能会得到衰老的生物标志物并推进旨在研发增殖和静态的衰老组织的康复治疗策略。这些治疗策略可能包括:通过瞬时激活端粒酶稳定端粒体;减少p53激活或者是与p53特异性竞争结合到衰老分子靶; 并增强PGC活性,促进线粒体生物合成和功能。沿着这些线路,已有报道端粒酶的小分子激活剂,在不增加癌症风险的前提下,并可延长雌性小鼠的健康寿命[24]。此外,骨骼肌中的PGC1α过表达,可改善野生型小鼠肌肉功能的年龄相关的退行性变[25]。
同样,已知提高线粒体生物合成和功能的其他干预,包括体力活动或服用白藜芦醇(推测为sirtuin的激活剂),也已被证实可以改善年龄相关的退行性变[26]。很有趣的是sirtuins可使P53活性降低和PGCs活性增加,去乙酰化活性降低,从而分别调制了连接DNA损伤信号和线粒体退行性变的通道中的两个关键组分。除外衰老,最近的研究还发现癌症中端粒-p53-线粒体轴的的重要性,这表明该通路可靶向用于癌症的治疗[27]。衰老的分子环路的发现为衰老定位了制定合理的策略,衰老是一类有100%外显率和100%死亡率的“疾病”。虽然仍有待确定是否可以阻止甚或逆转自然衰老,但是如最近的早老研究表明,衰老通路的干预性治疗可能有希望减少与年龄相关疾病,这将会促进全球范围内人类预期寿命明显增加。
参考文献
1Zoncu R, Efeyan A, Sabatini DM.mTOR: from growth signal integration to cancer, diabetes and ageing[J].Nature Rev Mol Cell Biol,2011,12:21-35.
2Hardie DG.Signal transduction: How cells sense energy[J].Nature.2011,472:176-177.
3Calado RT, Young NS.Telomere diseases[J].N Engl J Med,2009,361:2353-2365.
4Martin GM.Genetic modulation of senescent phenotypes in Homo sapiens[J].Cell,2005,120:523-532.
5Hewitt G, et al.Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence[J].Nature Commun,2012,3:708.
6Hao LY, et al.Short telomeres, even in the presence of telomerase, limit tissue renewal capacity[J].Cell,2005,123:1121-1131.
7Tomas-Loba A, et al.Telomerase reverse transcriptase delays aging in cancer-resistant mice[J].Cell,2008,135:609-622.
8Chang S, et al.Essential role of limiting telomeres in the pathogenesis of Werner syndrome[J].Nature Genet,2004,36:877-882.
9Jaskelioff M, et al.Telomerase reactivation reverses tissue degeneration in aged telomerasedeficient mice[J].Nature,2011,469:102-106.
10Sharpless NE, DePinho RA.Telomeres, stem cells, senescence, and cancer[J].J Clin Invest,2004,113:160-168.
11Choudhury AR, et al.Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation[J].Nature Genet,2007,39:99-105.
12Passos JF, et al.Feedback between p21 and reactive oxygen production is necessary for cell senescence[J].Mol Syst Biol,2010,6:347.
13Guo N, et al.Short telomeres compromise β-cell signaling and survival[J].PLoS ONE,2011,6:e17858.
14Vidal-Cardenas SL, Greider CW.Comparing effects of mTR and mTERT deletion on gene expression and DNA damage response: a critical examination of telomere length maintenanceindependent roles of telomerase[J].Nucleic Acids Res,2010,38:60-71.
15Sharma NK, et al.Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria[J].Nucleic Acids Res,2011,40:712-725.
16Pallardo FV, et al.Mitochondrial dysfunction in some oxidative stress-related genetic diseases: ataxia-telangiectasia, down syndrome, Fanconi anaemia and Werner syndrome[J].Biogerontology,2010,11:401-419.
17Varela I, et al.Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation[J].Nature,2005,437:564-568.
18Kim WY, Sharpless NE.The regulation of INK4/ARF in cancer and aging[J].Cell,2006,127:265-275.
19Ergün Sahin, Ronald A.DePinho.Axis of ageing: telomeres, p53 and mitochondria[J].Nat Rev Mol Cell Biol,2013,13(6): 397-404.
20Finkel T, Deng CX, Mostoslavsky R.Recent progress in the biology and physiology of sirtuins[J].Nature,2009,460:587-591.
21Burnett C, et al.Absence of effects of Sir2 overexpression on lifespan in C.elegans and Drosophila[J].Nature,2011,477:482-485.
22Passos JF, et al.Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomeredependent senescence[J].PLoS Biol,2007,5:e110.
23de Jesus BB, et al.The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence[J].Aging Cell,2011,10:604-621.
24Wenz T, Rossi SG, Rotundo RL, et al.Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging[J].Proc Natl Acad Sci USA,2009,106:20405-20410.
25Little JP, Safdar A, Bishop D, et al.An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1α and activates mitochondrial biogenesis in human skeletal muscle[J].Am J Physiol Regul Integr Comp Physiol,2011,300:R1303-R1310.
26Timmers S, et al.Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans[J].Cell Metab,2011,14:612-622.
27Hu J, et al.Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer[J].Cell,2012,148:651-663.
作者单位:1.佳木斯大学附属第一医院肿瘤科145002
基金项目:国家自然科学基金(81061120527,81370445,81472408);卫生部公益性研究基金(201302008);
doi:10.3969/j.issn.1672-4860.2015.04.001
收稿日期:2015-6-18
The panorama views of aging degeneration research(CHENFujun1,YANGZe2.1.ThefirsthospitalofJiamusiuniversity,Jiamusi145002.2.Beijinghospital,Beijing100730,China.)
【Abstract】Recent studies have found some new aging mechanisms mediated by telomere ie, telomere shortening and related DNA damage response, which will lead to mitochondrial dysfunction occurrence, reduce defense of oxidative damage, and undermine the process of ATP production, “different path…cross and converge to mitochondria model”.This may explain the general decline of energy generation, the phenomenon in stem cells, progenitor cells and after mitotic tissues.
【Key words】telomere, DNA damage response, mitochondrial dysfunction, aging
※为通讯作者
